

Full Length Research Article
α-globin gene 20.5kb deletion and triplication mutations among Palestinian patients with microcytic hypochromic anemia
Lamiaa Sobhi Saqer1*, FathElrahman Mahdi Hassan2, Fadel Akram Sharif3
Adv. life sci., vol. 12, no. 2, pp. 340-345, May 2025
*– Corresponding Author: Lamiaa Sobhi Saqer (Email: l.saqer@cst.ps)
Authors' Affiliations
2. Department of Hematology and Immunohematology, Collage of Medical Laboratory Science, Sudan University of Science and Technology – Sudan
3. Islamic University of Gaza – Palestinian Territory, Occupied
[Date Received: 20/10/2024; Date Revised: 05/01/2025; Available Online:22/03/2025; Date Updated: 31/08/2025]
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: α0-thalassemias results from double gene deletions of the α-genes on the same chromosome as in (-α)20.5. In contrast to deletion, α-globin genes triplication (αααanti3.7) is generated by homologous recombination during the process of crossover. This study was conducted to detect (-α)20.5 and ααα anti3.7 mutations in patients with microcytic hypochromic anemia in Gaza Strip-Palestine.
Methods: 200 subjects with microcytic hypochromic anemia with an age range of 18 to 48 years, were recruited from hematological departments of the main hospitals in Gaza Strip. The study subjects were those who underwent premarital β-thalassemia carrier screening, and their results were negative. Iron deficiency was excluded through measurement of serum iron and total iron binding capacity (TIBC). Complete blood count was done automatically. Molecular detection of (-α)20.5 and αααanti3.7 mutations were performed by Multiplex-PCR.
Results: Sixty-six (33%) of the study participants proved to have α-thalassemia. The frequency of (-α)20.5 and αααanti3.7 mutations were: 13.25% and 5.5%, respectively. Four genotypes were detected: –α20.5/αα accounted for 26% (52/200), αααanti3.7/αα for 2.5% (5/200), αααanti3.7/αααanti3.7 for 4% (8/200) and αααanti3.7/–α20.5 for 0.5% (1/200). The comparison of hematological parameters between –α20.5/αα and αααanti3.7 mutation carriers (αααanti3.7/αα and αααanti3.7/αααanti3.7) revealed, though not statistically significant, higher levels in carriers of αααanti3.7 mutation. The –α20.5/αααanti3.7 genotype was observed in one case.
Conclusion: The rate of –α20.5 and αααanti3.7 in the Palestinian population is high as compared to neighboring countries. Conducting further molecular testing to detect additional α-thalassemia mutations is needed in order to obtain a clearer picture of the genetic nature of this disease.
Keywords: α-thalassemia; –α20.5 deletion; αααanti3.7 triplication; Multiplex-PCR; Gaza Strip
Introduction![]()
α0-thalassemia deletion is caused by complete or partial deletion of both α-genes in cis thus eliminating the dictated α-chain synthesis. Homozygotes for such deletions have the Hb Bart’s Hydrops Fetalis Syndrome. There are about 20 α0 thalassemia deletions that tend to be frequent in various populations, four of which are more commonly found: —Med (Mediterranean) and
(-α)20.5 deletions in Mediterranean populations, and —SEA (South Asia) and —FIL (Filipino) in Southeast Asia. The breakpoints of the common α0 thalassemia deletions are caused by non-homologous recombination [1]. -(α)20.5 deletion, an α0-deletion commonly found in the Mediterranean and Central Asian populations. This deletion is reported to span 20.5 kb on the α-globin gene cluster, the 5' and 3' breakpoints are localized within an Alu region between the HBZ and HBZP1 genes and the HBA1 gene, respectively [2].
α3.7 triplication is an increase in α gene copies that occur on one of the chromosomes. α3.7 triplication is generated in a process called unequal crossover during the recombination of α1 and α2 genes, which creates the αααanti3.7 triplicated allele [3]. The mutual ααα anti3.7 allele of this unequal crossover consequently contains three α- globin genes because of the addition of a hybrid α-globin gene consisting of the 5′ portion of HBA1 and the 3′ portion of HBA2. Because a single-gene deletion and its corresponding triplication are mutual products of unequal crossover, equal occurrences of these alleles would be probable under the assumption of evolutionary neutrality for both alleles. In sperm, a high rate of mutual unequal crossover of the α-globin genes has been recognized, with equal occurrences of the α3.7 deletion and αααanti3.7 triplication [4]. However, between these mutual alleles, the α3.7 chromosomes are more common than the αααanti3.7 chromosomes, given a strong indication of positive environmental selection for the single-α-globin gene deletion in certain populations [5,6]. To our knowledge, this is the first study in the Gaza Strip investigating the α-globin gene mutations. In this study, multiplex PCR was used to identify the α-thalassemia 20.5 double deletion and ααα3.7 triplication mutations among Palestinian patients suffering from microcytic hypochromic anemia.
Methods![]()
The current analytical cross-sectional study employed two hundred microcytic hypochromic anaemic patients with MCV <80fL and/or MCH <27 pg collected from three hospitals in Gaza Strip: Al-Shifa, Gaza-European and Nasser Medical complex. All participants underwent β-thalassemia carrier screening premarital, and their results were negative. To exclude iron deficiency anemia; serum iron and TIBC were measured. Complete blood count was performed by an automated cell counter (XT1800i, Sysmex, Japan). All data were analyzed by SPSS version 20 using Independent Samples t-test and ANOVA for comparison of the hematological data. P value < 0.05 was considered statistically significant. The experimental work was conducted at the Labs of Medical Science department at the University College of Science and Technology – Gaza Strip, Palestine.
Ethical Considerations
An authorization to carry out the study was obtained from the Palestinian Ministry of Health and the Helsinki committee. In addition, all the subjects involved in this study gave their oral consent to participate in the study.
Detection of the α thalassemia mutations
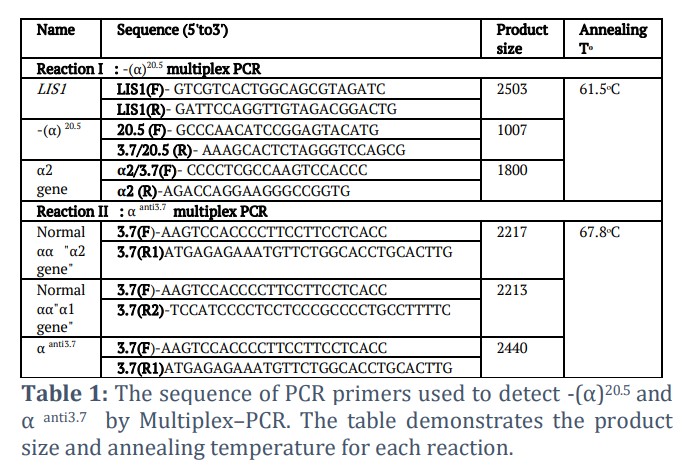
DNA was isolated from fresh EDTA whole blood cells by using Promega kit for human DNA isolation (Promega, USA). The quality and concentration of isolated DNA were determined by using Nanodrop spectrophotometer (IMPLEN-USA). One µl of the isolated DNA of each sample was used for this purpose. Multiplex-PCR was performed using the primers listed in Table 1.
Two micro-tubes were used: for reaction I -(α)20.5 multiplex PCR [7]: we used 15 μL master mix (Bioline), 2 μL deionized water, 1 μL DNA template, and 0.2 μL of each primer listed in Table 1 for αᵒ multiplex PCR (2 pmol) in one micro-tube (0.2 ml) and were mixed. In the second micro-tube used for reaction II- α anti3.7multiplex PCR [8], 7 μL master mix (Bioline), 2 μL deionized water, 1 μL DNA template, and 0.3 μL of forward primer (3 pmol) and 0.1 μL of each reverse primer in one micro-tube (0.2 ml) were mixed. Amplification of a large (2.5 kilobase) segment of the LIS1 gene 3'UTR (the LIS1 gene at 17p13.3) was included as internal control for monitoring the success amplification of reaction I. The sequence of primers used annealing temperature, and the PCR amplicons sizes are demonstrated in table1.
The thermal cycler (BECO-Germany) program was set as follows: initial denaturation at 95 °C for 3 min, followed by 25 cycles of denaturation at 95 °C for 30 s, annealing at 61.5 °C (reaction I) or 67.8 °C (reaction II) for 4 min, followed by an extension step at 72 °C for 4 min, and a final elongation at 72 °C for 5 min terminated the PCR reactions. A 1.5% agarose gel stained with ethidium bromide was used to detect the PCR products and visualized on a UV transilluminator. We estimated the PCR products sizes by comparison with 1kb ladder DNA run on the same gel.
Results![]()
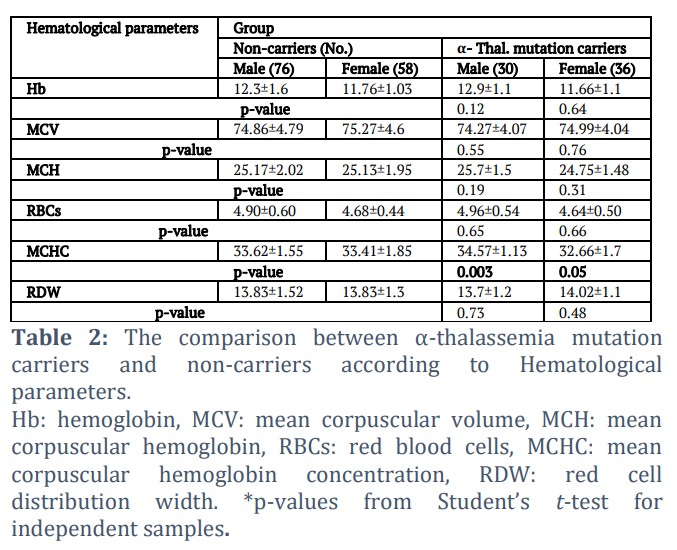
The study population consisted of 200 participants: 106 were males and 94 females, the participant's age ranged from 18 to 48 with an average of 30.35±9.6 years. All of the participants were microcytic hypochromic anemic patients with MCV: 74.9fl ±4.4 and/or MCH 25.16 pg ±1.8. Iron deficiency was excluded as a cause of their anemia by measuring their serum iron and TIBC. All the subjects underwent premarital carrier screening for β-thalassemia and their results came out negative. Among the 200 participants, 66 (33%) proved to be carriers for α-thalassemia based on the molecular analysis. An assessment of the hematological parameters among the mutation carriers and non-carriers are summarized in Table 2. The comparison showed no significant difference (p˃ 0.05) in terms of MCV, hemoglobin, RBCs, and RDW, regardless of gender. But the difference in the mean MCHC reached significance in α- thalassemia mutation carriers when compared with non-carriers with p-values: 0.05 and 0.003 for females and males, respectively.
Hb: hemoglobin, MCV: mean corpuscular volume, MCH: mean corpuscular hemoglobin, RBCs: red blood cells, MCHC: mean corpuscular hemoglobin concentration, RDW: red cell distribution width. *p-values from Student’s t-test for independent samples.
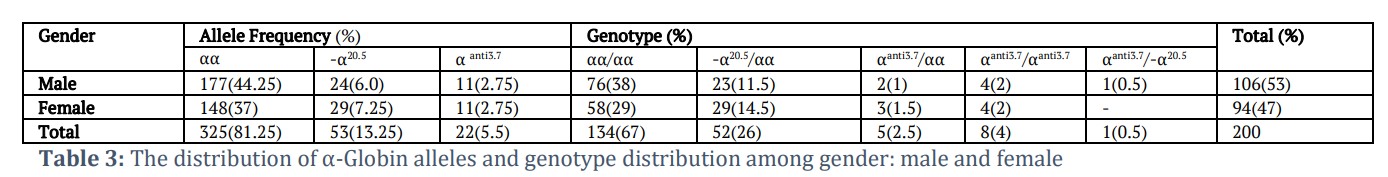
Of the two hundred DNA samples analyzed, 33% have been observed to be carriers of α-gene defect. Based on the molecular investigation, the mutations of interest (–α20.5) deletion and triplicated (αααanti3.7) were observed. The mutant alleles represented 18.75% of total α– globin alleles (mutant and normal alleles). The frequency of –α20.5 mutation was (13.25%) while ααα anti3.7 (5.5%) Table (3). The frequencies of the –α20.5 deletion was higher in female (7.25%) than in male (6.0%) subjects. The frequencies of αααanti3.7 triplication, however, were the same in both genders (2.75%).
Among the α-thalassemia gene mutation carriers, four different α-globin genotypes were identified: –α20.5/αα (26%), αααanti3.7/αα (2.5%), αααanti3.7/αααanti3.7 (4%) and αααanti3.7/–α20.5 (0.5%).
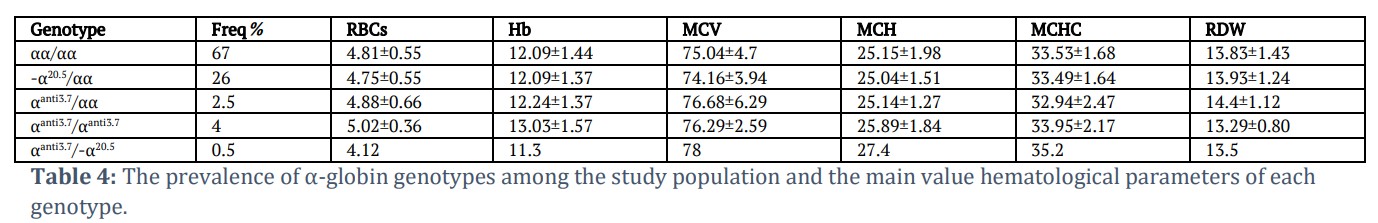
According to the results shown in table 4, the highest levels of RBCs and Hb were found in the αααanti3.7/αααanti3.7genotype [mean±SD:5.02±0.36 and 13.03±1.57], respectively. Whereas the lowest levels of MCV and MCH were seen in the -α20.5/αα (MCV:74.16fl and MCH: 25.04pg).As for αααanti3.7/αααanti3.7genotype the level of MCV was slightly decreased but the mean of RBCs, Hb and MCH levels were higher than in αααanti3.7/αα genotype; however, when combined with –α20.5 deletion (seen in one case) the levels of MCV and MCH were increased while the Hb and RBCs decreased (Table 4).
The rate of mutation occurrence in females was higher than that in males: 36/66 women (54.54%) and 30/66 men (45.45%). In the heterozygous group: 26/30 men (86.67%) and 32/36 women (88.88%), whereas 4/30 men (13.33%) and 4/36 women (11.11%) were in the homozygous group.
However, the combination –α20.5 and αanti3.7 was observed in only one male. Regarding the –α20.5 mutation; we observed that MCH levels and RDW were almost equal in the –α20.5/αα genotype when compared with normal genotype, however the mean of MCV levels and RBCs count were lower in –α20.5/αα genotype ; but these differences did not reach statistical significance. In contrast all the hematological parameters, except MCH and RDW, were elevated in the αααanti3.7 homozygotes as compared to the heterozygotes.
The highest levels of Hb, RBCs, MCV and MCH were observed in the αααanti3.7/αα and the αααanti3.7/αααanti3.7 genotypes.
Figures & Tables
![]() The most frequent mutation in our population –α20.5 was observed in 53 out of 200 patients and represented a frequency of 26.5%. This mutation is not often reported in Arab countries, the Middle East, and Asia. It was reported to be the less frequent deletion in the studies conducted by Mesbah-Amroun et al., [9] in Algeria (2008) with 6.5%, 8.9% in Saudi population [10] and 9% in the United Arab Emirates [11].
The most frequent mutation in our population –α20.5 was observed in 53 out of 200 patients and represented a frequency of 26.5%. This mutation is not often reported in Arab countries, the Middle East, and Asia. It was reported to be the less frequent deletion in the studies conducted by Mesbah-Amroun et al., [9] in Algeria (2008) with 6.5%, 8.9% in Saudi population [10] and 9% in the United Arab Emirates [11].
This mutation was detected in Sulaimani region in Iraq by Amin et al[12] (9.8%) but was not detected in Erbil province [13] in contrast Al-Allawi et al [14] identified this mutation by 2.9% in Northeastern Iraq. On the other hand, this mutation was not observed in Lebanon, Jordan or Tunisia in the studies conducted by Farra et al.[15], Qaddoumi et al [16] and Zorai et al.[17] respectively.
Pouranfard et al. [18] reported the –α20.5 mutation in 0.6% in a cross-sectional study carried out in a province located southwest of Iran. Our results differ from those reported from other neighboring Mediterranean such as Greece (12.7%), Cyprus (8.3%) and Turkey (5.8%) [19-21]. The high frequency of this mutation in our population may be due to the frequent consanguineous marriage.
In our study, the –α20.5 deletion was found in fifty-two cases as –α20.5/αα and one case was compound heterozygous with the αααanti3.7 triplicated allele.α-globin gene triplication is, actually, an increase in α-genes that occurs on one of the chromosomes. α-gene triplication (αααanti3.7) mechanism is an uneven crossover during the recombination of α1 and α2 gene [22-25]. If the crossover occurs between the homologous Z2 and Z1 boxes, also called a “rightward crossover”, then -α3.7 single-gene deletion allele and the mutual αααanti3.7 triplicated allele result.
There are various studies which displayed the frequency of the α-globin gene triplication in healthy subjects and thalassemia patients. The incidence of the α-globin gene triplication is diverse, and it is reliant on the prevalence of thalassemia disease in the studied countries [26]. There are two types of triplicated α-globin genes: αααanti3.7 and αααanti4.2 [27,28]. The αααanti4.2 is usually detected in Asians whereas the αααanti3.7 is predominant in Africans, Middle Eastern, and Mediterranean populations [26].In our study, we found heterozygous triplications αααanti3.7 in 5 cases and homozygous triplication in 8 cases and one subject carried a αααanti3.7 allele and the –α20.5allele (genotype: αααanti3.7/-α20.5). The αααanti3.7 allele accounted for 5.5% of the total α-globin alleles (22/400). All 14 carriers of the triplicated α-globin genes including the αααanti3.7/-α20.5 case did not present any clinical manifestations except microcytic hypochromic anemia at the time of examination. The blood parameters including the RBCs, Hb, MCV, MCH, MCHC and RDW were measured and statistically analyzed through ANOVA.
No significant parameter difference was identified among the different genotypes in comparison with the non-carrier subjects (p > 0.05) (data not shown). We observed that the higher mean of Hb level and RBCs was in αααanti3.7/ αααanti3.7 genotype among the three different genotypes. αααanti3.7/-α20.5genotype has the lowest Hb level (11.3g/dl) and RBCs count beside the highest value of MCV and MCH among the 3 genotypes. It appears that the extra α-gene in the case of the αααanti3.7 triplication mutation replaces the decrease caused by the deletion of the α-globin gene due to the –α20.5 mutation.
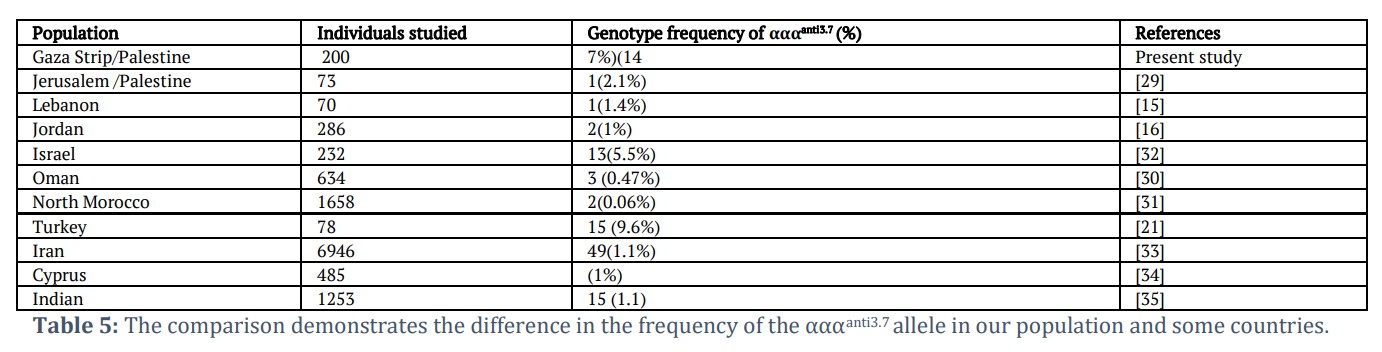
In our study fourteen cases were found to have αααanti3.7 triplication (29.33% of mutant chromosome:22/75) while this mutation was reported in 2.7 % of mutant chromosomes in a study conducted by Hamayel et al. in Jerusalem/Palestine[29]. Various studies identified the αααanti3.7 triplication: Lebanon [15], Jordan [16], Oman [30], North Morocco [31], Turkey [21] and others (Table 5).
It should be noted that the frequency of the α-thalassemia carriers should be higher in Gaza Strip because in this study only microcytosis and hypochromia cases were included in this study, so the prevalence of the α-thalassemia carriers might be underestimated and only two mutations have been investigated in our study population.
Still, the –α20.5 deletion with a frequency of 13.25% and the αααanti3.7 triplication with a frequency of 5.5% constitute important α-thalassemia mutations in the investigated population and should be considered when investigating microcytic hypochromic patients.
Author Contributions
Lamiaa Sobhi Saqer carried out the experiment and wrote the manuscript. FathElrahman Mahdi Hassan and Fadel Akram Sharif verified the analytical methods and supervised the findings of this work. All authors discussed the results and contributed to the final manuscript.
The authors declare that there is no conflict of interest.![]()
References
- Traeger-Synodinos J . Molecular basis of α-thalassaemia. Thalassemia Reports, (2011); 1(2): 48-51.
- Souza AE, Cardoso GL, Takanashi SY, Guerreiro JF. α-Thalassemia (3.7 kb deletion) in a population from the Brazilian Amazon region: Santarém, Pará State. Genetics and Molecular Research, (2009); 8(2): 477-481.
- Hamid M, Keikhaei B, Galehdari H, Saberi A, Sedaghat A, Shariati G, et al. Alpha-globin gene triplication and its effect in beta-thalassemia carrier, sickle cell trait, and healthy individual. Journal of Haematology, (2021); 2(3): 366-374.
- Lam KW, Jeffreys AJ . Processes of de novo duplication of human alpha-globin genes. Proceedings of the National Academy of Sciences, (2007); 104(26): 10950-10955.
- Weatherall DJ. Pathophysiology of thalassaemia. Baillieres Clinical Haematology, (1998); 11(1): 127-146.
- Goossens M, Dozy AM, Embury SH, Zachariades Z, Hadjiminas MG, Stamatoyannopoulos G, et al. Triplicate alpha-globin loci in humans. Proceedings of the National Academy of Sciences, (1980); 77: 518-521.
- Chong SS, Boehm CD, Cutting GR, Higgs DR. Simplified multiplex-PCR diagnosis of common southeast Asian deletional determinants of alpha-thalassemia. Clinical Chemistry, (2000); 46(10): 1692-1695.
- Liu YT, Old JM, Miles K, Fisher CA, Weatherall DJ, Clegg JB. Rapid detection of alpha-thalassaemia deletions and alpha-globin gene triplication by multiplex polymerase chain reactions. British journal of haematology, (2000); 108(2): 295-299.
- Mesbah-Amroun H, Rouabhi F, Ducrocq R, Elion J. Molecular basis of alpha-thalassemia in Algeria. Hemoglobin, (2008); 32: 273-278.
- Akhtar MS, Qaw F, Borgio JF, Albuali W, Suliman A, Suliman A, et al. Spectrum of α-thalassemia mutations in transfusion-dependent β-thalassemia patients from the Eastern Province of Saudi Arabia. Hemoglobin, (2013); 37(1): 65-73.
- Baysal E. α-Thalassemia Syndromes in the United Arab Emirates. Hemoglobin, (2011); 35(5-6): 574-580.
- Amin LN, Rasool LK, Nore BF, Salih GF . Description of Hemoglobin H disease mutation in alpha thalassemia patients in Sulaimani region in Kordistan region ,Iraq. Iraqi Journal of hematology, (2021); 10(2): 97-101.
- Shamoon RP. Molecular spectrum of α‑thalassemia mutations in Erbil province of Iraqi Kurdistan . Molecular Biology Reports, (2020); 47(8): 6067-6071.
- Al-Allawi N, Jalal SD, Rasheed NS, Bayat N, Imanian H, et al. The spectrum of α-thalassemia mutations in the Kurdish population of Northeastern Iraq. Hemoglobin, (2013); 37(1): 56-64.
- Farra C. Daher R, Badra R, Rafei R, Bejjany R, Charafeddine L, et al. Incidence of Alpha-Globin Gene Defect in the Lebanese Population: A Pilot Study. BioMed Research International, (2015); 2015: 1-3.
- Qaddoumi A, Kamal N, Shbailat T. Molecular Spectrum of Alpha-Thalassemia in Jordan. Journal of The Royal Medical Services, (2008); 15(2): 23-27.
- Zorai A, Abbes S, Préhu C, Omar S, Gerard N, et al. Hb H disease among Tunisians: molecular characterization of α-thalassemia determinants and haematological findings. Hemoglobin, (2003); 27(1): 57-61.
- Pouranfard J, Vafaei F, Afrouz S, Rezaeian M .Thalassemia Gene Mutations in Kohgiluyeh and Boyer-Ahmad Province. Iranian Journal of Blood & Cancer, (2020); 12(1): 18-23.
- Kanavakis E, Papassotiriou I, Karagiorga M, Vrettou C, Metaxotou-Mavrommati A, Stamoulakatou A, et al. Phenotypic and molecular diversity of haemoglobin H disease: a Greek experience. British Journal of Haematology, (2000); 111(3): 915-923.
- Baysal E, Kleanthous M, Bozkurt G, Kyrri A, Kalogirou E, Angastiniotis M, et al. alpha-Thalassaemia in the population of Cyprus. British Journal of Haematology, (1995); 89(3): 496-499.
- Demir S, Gürkan H, Eker D, Yalçıntepe S, Atlı EI, Atli E. Retrospective analysis of alpha globin copy number variations determined by MLPA in the Trakya region. Journal of Istanbul Faculty of Medicine, (2021); 84(3): 348-353.
- Ho PJ, Hall GW, Luo LY, Weatherall DJ, Thein SL. Beta-thalassaemia intermedia: is it possible consistently to predict phenotype from genotype?. British Journal of Hematology, (1998); 100(1): 70-78.
- Traeger –Synodinos J, Kanavakis E, Vrettou C, Maragoudaki E, Michael T,Metaxotou-Mavromati A, et al. The triplicated alpha-globin gene locus in beta thalassaemia heterozygotes: clinical, haematological, biosynthetic and molecular studies. British Journal of Hematology, (1996); 95(3): 467-471.
- Camaschella C, Kattamis AC, Petroni D, Roetto A, Sivera P, et al. Different hematological phenotypes caused by the interaction of triplicated α-globin genes and heterozygous β-thalassemia. American Journal of Hematology, (1997); 55(2): 83-88.
- Colah RB, Nadkarni AH, Mukherjee MB, Gorakshakar AC, Surve R, Mohanty D. Beta thalassaemia heterozygotes with alpha-globin gene triplication. British Journal of Hematology, (1997); 97(2): 506-507.
- Giordano PC, Bakker-Verwij M, Harteveld CL. Frequency of alpha-globin gene triplications and their interaction with beta-thalassemia mutations. Hemoglobin, (2009); 33(2): 124-131.
- Lie-Injo LE, Herrera AR, Kan YW. Two types of triplicated alpha-globin loci in humans. Nucleic Acids Research, (1981); 9(15): 3707-3717.
- Trent RJ, Higgs DR, Clegg JB, Weatherall DJ. A new triplicated alpha-globin gene arrangement in man. British Journal of Hematology, (1981); 49(1): 149-152.
- Hamayel, M. Genotyping of Alpha-Globin Gene Mutations among Palestinian Patients with Unexplained Microcytosis. The Al-Quds University, (2013). Available at: https://dspace.alquds.edu/handle/20.500.12213/1621.
- Hassan M, Harteveld CL, Bakker E, Giordano PC. Molecular spectrum of α-globin gene defects in the Omani population. Hemoglobin, (2014); 38(6): 422-426.
- Laghmich A, Alaoui Ismaili FZ, Barakat A, Ghailani Nourouti N, Khattab M, Bennani Mechita M. Alpha-Thalassemia in North Morocco: Prevalence and Molecular Spectrum. BioMed Research International, (2019); 2019: 1-7.
- Oron-Karni V, ilon D, Shifrin Y, Fried E, Pogrebijsky G, Oppenhein A, et al. Diversity of α-globin mutation and clinical presentation of α-Thalassemia in Israel. American Journal of Hematology, (2000); 65(3): 196-203.
- Ebrahimi M, Mohammadi-asl J, Rahim F. Molecular spectrum and distribution of hemoglobinopathies in southwest of Iran: a seven-year retrospective study. Journal of Hematopathology, (2020); 13: 97-103.
- Hamamy HA, Al-Allawi NA. Epidemiological profile of common hemoglobinopathies in Arab countries. Journal of Community Genetics, (2013); 4(2): 147-167.
- Nadkarni A, Phanasgaonkar S, Colah R, Mohanty D, Ghosh K. Prevalence and molecular characterization of α-thalassemia syndromes among Indians. Testing, (2008); 12(2): 177-180.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0