

Full Length Research Article
Deleterious Nonsynonymous Single Nucleotide Polymorphisms (nsSNPs) in the Human Interleukin 12B Gene: Identification and Structural Characterization
Awad A. Algarni
Adv. life sci., vol. 10, no. 1, pp. 129-135, March 2023
*– Corresponding Author: Awad A. Algarni (Email: koroshs.saki@gmail.com)
Authors' Affiliations
Editorial Note on Version of Record
05 Sept 2025: This article has been corrected. See https://doi.org/10.62940/als.v13i0.4227 for more information.
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Interleukin-12B (IL12B) polymorphism has been identified as a factor in the development of various Immunological disorders and cancer. The objective of this study was to identify the non-synonymous SNPs (nsSNPs) with the strongest predicted negative impact on the function of the IL12B protein.
Methods: We employed a variety of computational methods, including SIFT, PolyPhen-2, PROVEAN, and SNAP2 to determine the functional impact of nsSNPs. Also, in order to investigate the potential association of nsSNPs in the IL12B gene with disease, a computational analysis was conducted using PhD-SNP, SNPs&GO, and PMut. Additionally, I-Mutant and MuPro were employed to predict protein stability, while ConSurf was used to identify functional domains and conserved amino acid residues within the protein. Furthermore, SOPMA was used in combination with Project Hope and MutPred2 to predict the impact of mutations on both the structure and function of proteins. Finally, we used GeneMania to analyze the gene-gene interactions of the IL12B gene with other genes.
Results: Our results indicate that nine nsSNPs (G72C, G86C, C90R, C131S, Y136D, P235L, V254G, Y258H and P259S) were found to be potentially deleterious in the IL12B gene.
Conclusion: Our study emphasizes the significance of identifying functional and structural polymorphisms in the IL12B gene, as they may reveal potential therapeutic targets and provide insight into the underlying mechanisms of related diseases. Further experimental investigation is necessary to fully explore the role of these nsSNPs in disease pathogenesis.
Keywords: Interleukin 12B; deleterious nsSNPs; Polymorphisms.; Computational analysis; Cancer
Introduction![]()
Interleukin-12B (IL12B) gene, located on chromosome 5q33.3, encodes the p40 subunit of the cytokines IL12 and IL23 [1]. These cytokines, produced by this gene, are vital for both innate and adaptive immunity. These cytokines are essential for activation, differentiation, and proliferation T-helper cells, which are central to the immune system’s response to infections. Additionally, IL-12 and IL-23 are responsible for activating natural killer cells and controlling the inflammatory response [2,3]. The IL12B polymorphism has been associated with the onset of various immune-related disorders, including type-1 diabetes [4], inflammatory bowel disease [5], asthma [6], rheumatoid arthritis [7], allergic rhinitis [8], and Alopecia areata [9]. Furthermore, multiple investigations utilizing both molecular and genetic epidemiological techniques have revealed that specific polymorphisms within the IL12A and IL12B genes may be correlated with an increased risk of certain cancer types, such as lung, breast, and gastric cancers [10-12]. The human genome exhibits a high degree of similarity at the global level, with 99.9% of DNA sequences being identical across all individuals. However, the remaining 0.1% of the genome is composed of genetic variations that arise from random mutations. The frequently observed form of genetic diversity is the single nucleotide polymorphism (SNP), which is characterized by the replacement of a single nucleotide at a specific locus within the genome [13]. SNPs represent a common form of genetic variation, with an estimated frequency of approximately 1 in every 1,000 base pairs in the genome. These variations are particularly prevalent within coding regions, which have a fundamental impact on the expression of genetic information [14]. A particular subclass of SNPs are the non-synonymous SNPs (nsSNPs) that lead to the replacement of amino acids and subsequent alteration of protein structure in humans. A significant body of literature has established that nsSNPs are a major contributor to mutations associated with a broad spectrum of genetic disorders, as well as several inflammatory and autoimmune disorders [15,16]. Currently, there has been no comprehensive computational analysis of the IL12B gene to identify all potential nsSNPs that could alter the activity or conformation of the protein. This study endeavors to utilize a range of in silico methods to analyze the impact of nsSNPs on the IL12B gene to determine any potential negative consequences. Instead of relying on experimental validation, the study aims to provide a rapid and cost-effective method for identifying nsSNPs that are pathogenic.
Methods![]()
Dataset Collection
Data on the IL12B gene was obtained from the NCBI. While the sequence of amino acids was obtained from the UniProt database, identified by its accession number P29460. This study focused on utilizing missense SNPs (nsSNPs) exclusively.
Functional deleterious nsSNPs analysis
To evaluate the functional consequences of nsSNPs, we employed a comprehensive analytical approach utilizing four distinct bioinformatic tools. The SIFT algorithm assesses the effect of changing one amino acid with another on the performance of a protein. It takes into consideration both the resemblance of the sequence and the chemical and physical properties of the substituted amino acid to make its assessment [17]. The PROVEAN is a method for identifying non-synonymous variants that may impact protein function. By using alignment-based scores, PROVEAN can determine whether an amino acid variation is likely to be detrimental or benign [18]. The PolyPhen-2 algorithm utilizes a combination of structural, sequence, and evolutionary information to anticipate the functional effects of altering amino acids in proteins [19]. PANTHER, on the other hand, employs evolutionary data to evaluate the potential effect of single nucleotide variations on proteins’ function [20].
Disease-associated nsSNPs analysis.
The Pmut server predicts the pathogenicity of mutations using a neural network algorithm. It does this by analyzing a dataset of manually curated genetic variations from the SwissVar database. This allows the server to accurately identify genetic variations associated with disease [21]. The PhD-SNP tool employs the support vector machine method to assess the association of a certain nsSNP with a pathological condition. The outcome of the prediction includes a reliability index, which reflects the probability that the SNP in question is linked to disease or is benign in nature [22]. A reliable machine learning technique, based on support vector machine method, known as SNPs&GO was employed to predict disease-associated amino acid variations in proteins through the integration of information derived from protein sequence and structure [23].
Protein stability analysis
In silico analysis was conducted using both I-Mutant 2.0 and MuPro tools to scrutinize the association of mutations and the structural integrity and performance of proteins.I-Mutant 2.0 analyzes how mutations affect the stability of proteins by determining the shift in unbound energy (ΔΔG). Negative values suggest that the mutation decreases stability and positive values indicate that the mutation increases stability [24]. MuPro evaluates the potential effect of single nucleotide variations on protein stability using only SVM and sequence information, by determining both the magnitude and direction of energy change [25].
Conservation analysis
ConSurf, a bioinformatics tool, is utilized to analyze the sequence of a protein and compare it to similar sequences in order to determine the extent to which certain amino acid residues have been conserved over evolutionary time. This can provide insights into the functional or structural importance of these residues in the protein. It assigns a conservation score to each residue, with higher scores indicating greater preservation [26].
Secondary Structures analysis
SOPMA is an algorithm that utilizes information about the location of amino acids in a protein to predict its secondary structure. It uses a self-optimized prediction method and considers multiple alignments of similar protein sequences when making its predictions [27].
Protein properties analysis
HOPE is a tool that automates the process of analyzing protein mutants. HOPE uses information from the UniProt database to analyze how point mutations affect the structural integrity and performance of proteins. It produces a detailed report that includes text, figures, and animations to show the impact of the mutations [28].
Molecular mechanism of pathogenicity analysis
MutPred is an online server that utilizes amino acid substitution in mutant proteins to predict the molecular basis of disease, it also screens for Modifications in protein conformation such as loss of catalytic sites, altered stability, and gain of O-linked glycosylation [29].
Gene-gene interaction analysis
GeneMANIA is a tool that predicts the functions of unidentified proteins by integrating data from proteomics and genomics from various sources [30].
Results![]()
SNP datasets
NCBI dbSNP reports that there are 3874 SNPs found in the IL12B gene. Among these, 284 SNPs are classified as missense mutations that were chosen for further examination.
Functional deleterious SNPs analysis
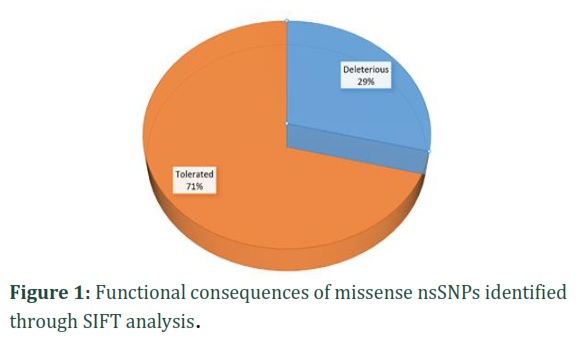
An analysis of the functional consequences of 284 missense nsSNPs was conducted using the SIFT server. The results of this study showed with high confidence that 82 nsSNPs were identified as deleterious, and 202 nsSNPs were identified as tolerated (Figure 1).To confirm these findings, the 82 missense nsSNPs identified as deleterious by SIFT were further analyzed using additional computational tools, including PolyPhen-2, PROVEAN, PANTHER, SNAP2.
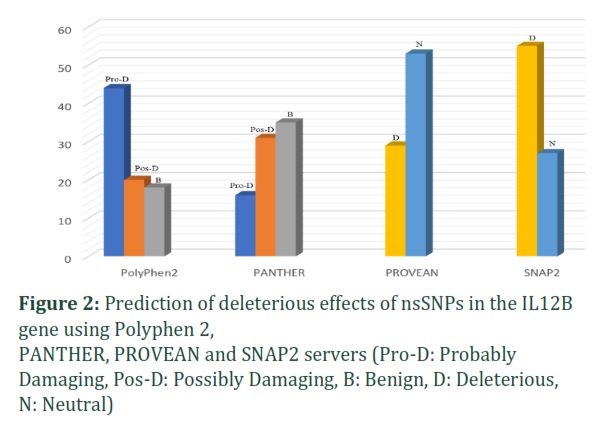
The PolyPhen-2 predicted that 44 nsSNPs were probably damaging, 20 nsSNPs were possibly damaging, and 18 nsSNPs were benign. The PROVEAN server predicted that 29 nsSNPs were deleterious and 53 were neutral. Meanwhile, the PANTHER server predicted that 16 nsSNPs were probably damaging, 31 nsSNPs were possibly damaging, and 35 SNPs were probably benign. The SNAP2 server predicted that 55 nsSNPs were deleterious and 27 were neutral. Out of 82 nsSNPs, 20 were identified as highly deleterious (Figure 2).
PANTHER, PROVEAN and SNAP2 servers (Pro-D: Probably Damaging, Pos-D: Possibly Damaging, B: Benign, D: Deleterious, N: Neutral)
Disease-associated nsSNPs analysis
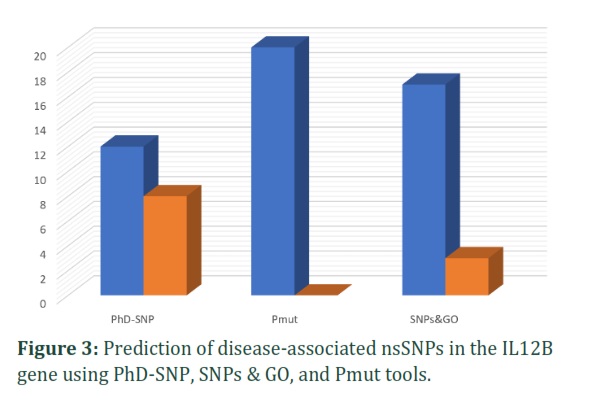
To acquire a deeper understanding into the disease-risk potential of nsSNPs, 20 nsSNPs were analyzed using a combination of PhD-SNP, Pmut, and SNP&GO. The PhD-SNP analysis revealed that 12 nsSNPs have a potential association with disease, while 8 nsSNPs were predicted to be neutral. The Pmut software identified 20 nsSNPs as having a potential association with disease and none as neutral. According to the SNPs&GO analysis, 17 nsSNPs were found to potentially be linked to disease, while 3 nsSNPs were deemed neutral. The analysis of the IL12B gene revealed that 11 nsSNPs were found to be associated with disease-associated variants (Figure 3). Given their classification as the most deleterious nsSNPs, these 11 nsSNPs were subject to further analysis to gain a more comprehensive knowledge of their potential influence.
Protein stability analysis
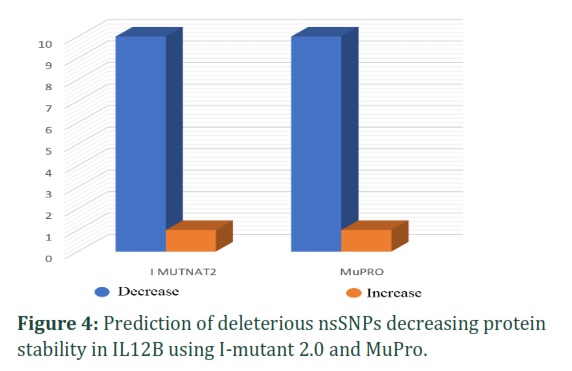
An analysis was conducted using I-mutant 2.0 and MuPro to evaluate the impact of 11 nsSNPs on the stability of a protein. The results of I-mutant 2.0 indicated that 10 nsSNPs (G72C, G86C, C90R, C131S, Y136D, F140L, P235L, V254G, Y258H, P259S) are likely to decrease protein stability, and MuPro produced similar results by predicting decreased stability for 10 nsSNPs (C50Y, G72C, G86C, C90R, C131S, Y136D, P235L, V254G, Y258H, P259S) (Figure 4).
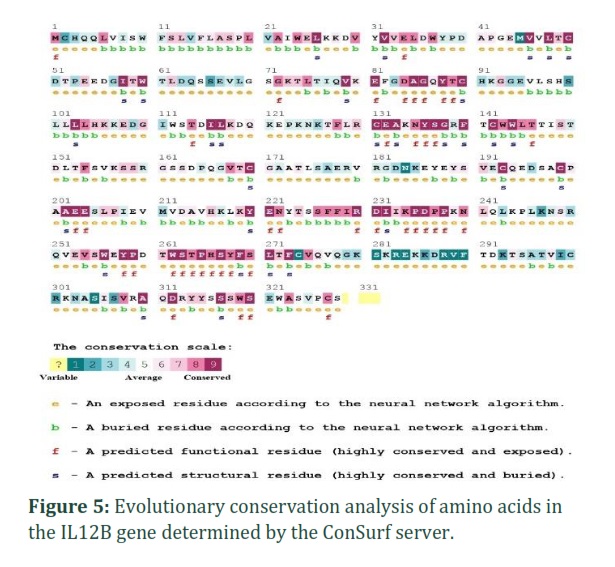
Conservation analysis
As shown in figure (5), all 11 nsSNPs were predicted as highly conserved using ConSurf server. The analysis revealed that C50Y, C90R, C131S, F140L, and V254G have high conservation scores of 9 and 8, being buried and structural residues, while G72C, G86C, Y136D, P235L, Y258H, P259S were identified as highly conserved, exposed, and functional residues.
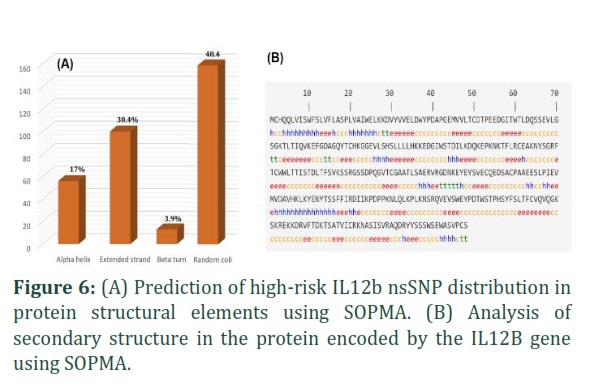
Secondary Structures analysis
SOPMA was used to analyze the secondary structures of IL12B protein. According to SOPMA, a total of 56 residues were found to be associated with alpha helix, accounting for 17.07% of the total. Additionally, 159 residues were identified as being part of a random coil, making up 48.1% of the residues. Extended strand was found to be present in 100 residues, constituting 30.49% of the total, and beta-turn was identified in 13 residues, representing 3.96% of the total. An analysis of the location of nsSNPs in secondary structures revealed that, among the deleterious nsSNPs, six were located in the random coil secondary structure (C50Y, G86C, Y136D, P235L, Y258H, P259S). Four of these deleterious SNPs were found in the extended strand (C90R, C131S, F140L, V254G) and only one (G72C) was in the β-turn. However, no deleterious nsSNPs were identified in the alpha helix (Figure 6).
Protein properties analysis
The HOPE method was applied to predict the outcome of 11 deleterious nsSNPs of the IL12B gene on various characteristics of the amino acids, including hydrophobicity, charge, size , and spatial structure function. Five mutant residues (C50Y, G72C, G86C, C90R, P235L) were identified as being larger in size compared to their wild residues. Moreover, five mutant residues (Y136D, F140L, V254G, Y258H, P259S) were determined to be smaller in size when compared to wild residues. In addition, HOPE revealed that two mutant residues, C90R and Y136D, experienced a change in charge. Specifically, C90R changed from a neutral to a positive charge, while Y136D changed from a neutral to a negative charge. Moreover, three of the mutant residues (C50Y, G72C, G86C) were more hydrophobic than the wild type and six (C90R, C131S, Y136D, V254G, Y258H, P259S) were less hydrophobic than the wild type.
Molecular mechanism of pathogenicity analysis
Figure (7) shows the probability scores for 11 nsSNPs that were submitted to MutPred2 server. The results of MutPred analysis indicate that many nsSNPs can lead to alterations in protein structure, such as changes in transmembrane regions, stability, metal binding sites, and ordered interfaces. Additionally, there are predictions of new features emerging, such as gain in strand, catalytic site, disulfide linkage, as well as loss of loop, sulfation, and disulfide linkage.
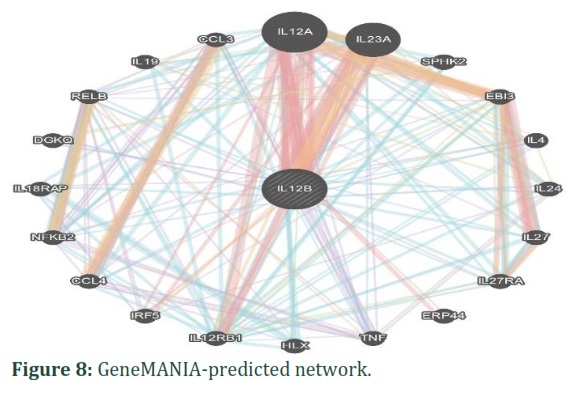
Gene-gene interaction analysis
IL12B gene interactions with other cellular genes were predicted using the GeneMANIA tool. Using GeneMANIA revealed predictions of physical interactions between IL12B and other genes, including IL12A, IL23A, ERP44, and IRF5.Five genes were identified as being predicted to have co-expression with IL12B: IL4, TNF, CCL4, DGKQ, and IL18RAP.In predicted, IL12B showed a relation with IL23A, IL12RB1 and IRF5.Co-localization analysis revealed a correlation between IL12B and multiple other genes, including IL12A, IL23A, IL4, IL24, TNF, and DGKQ. In genetic interactions, IL12B showed a relation with IL12A, HLX and IL12RB1.In pathways, IL12B showed a relation with IL12A, IL23A ,SPHK2, EBI13, IL4, IL24, IL27RA, TNF, HLX, IL12RB1, CCL4, NFKB2, IL18RAP, RELP,IL19 and CCL3. Finally, GeneMANIA analysis found that IL12B share a domain with EB13. The predictions generated by GeneMANIA are presented in figure 8, providing a comprehensive overview of the relationships between various genes.
Figures & Tables

The focal point of this investigation was to analyze the effects of nsSNPs in the human IL12B gene on protein functionality, stability, and structure by using a variety of bioinformatic tools. The methods employed in this study provide a molecular-level understanding of the effects of variations by analyzing various aspects and parameters related to the pathogenicity of specific amino acid substitutions. Given that different algorithms utilize distinct sets of sequences for alignments, the prediction capabilities of different methods may vary. Therefore, it is not reliable to rely solely on a single bioinformatic tool for predicting the pathogenic effects of nsSNPs. In this study, multiple methods were utilized to evaluate the impact of nsSNPs[31]. From the NCBI dbSNP database, we retrieved 3874 SNPs for the IL12B gene, out of which 284 were found to be missense SNPs.The functional impact of nsSNPs on IL12B gene was assessed using five computational prediction tools (SIFT, PolyPhen-2, PROVEAN, SNAP2, and Panther) and the potential association of nsSNPs with disease was predicted using three other tools (PhD-SNP, Pmut, and SNP&GO) resulting in the identification of 11 nsSNPs as potentially “high-risk” and deleterious.These 11 nsSNPs were used in further prediction assays. The protein stability of 11 high-risk nsSNPs using I-mutation2 and MuPro showed that 10 nsSNPs resulted in a decreasein protein stability. Phylogenetic analysis discovered that all the nsSNPs were present in areas with high conservation scores. It is likely that variations in these regions will have a significant impact the function of the protein [32]. The examination of the location of deleterious nsSNPs in secondary structures by SOPMA revealed that a majority of them were in the regions of random coil and extended strand.The analysis performed by HOPE found that genetic mutations can have a detrimental impact on protein function, manifesting as either a reduction in protein-protein interactions or structural changes, particularly in transmembrane regions. Additionally, the study found that variations in charge or hydrophobicity can result in repulsion between amino acids, improper folding, or a decline in interactions [33]. Furthermore, molecular mechanism of pathogenic analysis using MutPred2 revealed that all 11 nsSNPs cause altered transmembrane in IL12B protein. Also, MutPred2 results predicted that 3 nsSNPs (C90R, C131S, V254G) change the protein stability. Moreover, C90R and P235L showed a loss of loops, which can alter protein intrinsic functions [34]. The result predictions for (C50Y, C90R) indicated that these mutations may lead to an altered metal binding. It was also predicted that the (C50Y, C131S, P235L) and (C131S, P235L) would result in a gain of strand and an alteration of the ordered interface, respectively. An analysis of the gene-gene interactions of the IL12B was conducted using the GeneMANIA tool. The results indicated that the IL12B gene was found to have interactions with a variety of interleukins, including IL12A, IL23A, and IL4, as well as with the TNF and IRF5 genes.In conclusion, the current study determined that nine nsSNPs (G72C, G86C, C90R, C131S, Y136D, P235L, V254G, Y258H, P259S) in the IL12B gene were predicted to be the most deleterious and functionally significant. These nsSNPs can be employed in subsequent experimental studies to investigate their role in the related diseases.
![]()
References
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nature Reviews Immunology, (2003); 3(2):133-146.
- Moschen AR, Tilg H, Raine T. IL-12, IL-23 and IL-17 in IBD: immunobiology and therapeutic targeting. Nature Reviews Gastroenterology & Hepatology, (2019); 16(3): 185-196.
- Saravia J, Chapman, NM. and Chi H.. Helper T cell differentiation. Cellular & Molecular Immunology, (2019); 16(7): 634-643.
- Li J, Zhang C, Wang JB, Chen SS, Zhang TP, et al. Relationship between the IL12B (rs3212227) gene polymorphism and susceptibility to multiple autoimmune diseases: a meta-analysis. Modern Rheumatology, (2016); 26(5): 749-756.
- Wu PB, Wu XM, Qian R, Hong C, Yitian G, et al. Association between IL12B polymorphisms and inflammatory bowel disease in Caucasian population: a meta-analysis. Cytokine, (2020); 136:155296.
- Epaneshnikova VB, Smolnikova MV, Smirnova SV. Analysis of polymorphisms of genes regulating the immune response in patients with atopic asthma. Medical Genetics, (2016); 15(4):36-38.
- Manolova I, Ivanova M, Vasilev G, Stoilov R, Miteva L, Stanilova S. Impact of IL12B polymorphisms on genetic susceptibility and IL-12p40 and IL-23 serum levels in rheumatoid arthritis. Immunological Investigations, (2020); 49(1-2):1-4.
- Falahi S, Salari F, Rezaiemanesh A, Mortazavi SH, Koohyanizadeh F, et al. Association of interleukin-12B rs6887695 with susceptibility to allergic rhinitis. Immunologic Research, (2021); 69:189-195.
- Tabatabaei-Panah PS, Moravvej H, Delpasand S, Jafari M, Sepehri S, et al, Akbarzadeh R. IL12B and IL23R polymorphisms are associated with alopecia areata. Genes & Immunity, (2020); 21(3):203-210.
- Zhang W, Dang S, Zhang G, He H, Wen X. Genetic polymorphisms of IL-10, IL-18 and IL12B are associated with risk of non-small cell lung cancer in a Chinese Han population. International Immunopharmacology, (2019); 77:105938.
- Núñez-Marrero A, Arroyo N, Godoy L, Rahman MZ, Matta JL, Dutil J. SNPs in the interleukin-12 signaling pathway are associated with breast cancer risk in Puerto Rican women. Oncotarget, (2020); 11(37):3420
- Orenay-Boyacioglu S, Kasap E, Yuceyar H, Korkmaz M. Association of interleukin 12B rs3212227 polymorphism with gastric cancer, intestinal metaplasia, and helicobacter pylori infection. Genetika, (2020); 115-126.
- Emadi E, Akhoundi F, Kalantar SM, Emadi-Baygi M. Predicting the most deleterious missense nsSNPs of the protein isoforms of the human HLA-G gene and in silico evaluation of their structural and functional consequences. BMC genetics, (2020); 21(1):1-27
- Molineros JE, Looger LL, Kim K, Okada Y, Terao C, et al. Amino acid signatures of HLA Class-I and II molecules are strongly associated with SLE susceptibility and autoantibody production in Eastern Asians. PLoS genetics, (2019); 15(4):e1008092
- Ahmad HI, Ijaz N, Afzal G, Asif AR, Rahman A, et al. Computational Insights into the Structural and Functional Impacts of nsSNPs of bone morphogenetic proteins. BioMed Research International, (2022); 2022
- Allemailem KS, Almatroudi A, Alrumaihi F, Almansour NM, Aldakheel FM, et al. Single nucleotide polymorphisms (SNPs) in prostate cancer: its implications in diagnostics and therapeutics. American Journal of Translational Research, (2021); 13(4):3868.
- Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nature Protocols, (2016); 11(1):1-9
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE, (2012); 7:e46688.
- Poon KS. In silico analysis of BRCA1 and BRCA2 missense variants and the relevance in molecular genetic testing. Scientific Reports, (2021); 11(1):1-8.
- Dalmer TR, Clugston RD. Gene ontology enrichment analysis of congenital diaphragmatic hernia-associated genes. Pediatric Research, (2019); 85(1):13-19.
- López-Ferrando V, Gazzo A, De La Cruz X, Orozco M, Gelpí JL. PMut: a web-based tool for the annotation of pathological variants on proteins, 2017 update. Nucleic Acids Research, (2017); 45(W1):W222-W228.
- Capriotti E, Fariselli P. PhD-SNPg: a webserver and lightweight tool for scoring single nucleotide variants. Nucleic Acids Research, (2017); 45(W1):W247-52.
- Manfredi M, Savojardo C, Martelli PL, Casadio R. E-SNPs&GO: embedding of protein sequence and function improves the annotation of human pathogenic variants. Bioinformatics, (2022); 38(23):5168-5174.
- Capriotti E, Fariselli P, Casadio R. I-Mutant2. 0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Research, (2005); 33:W306-W310.
- Cheng J, Randall A, Baldi P. Prediction of protein stability changes for single‐site mutations using support vector machines. Proteins: Structure, Function, and Bioinformatics, (2006); 62(4):1125-1132.
- Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Research, (2010); 38:W529-W533.
- Deléage G. ALIGNSEC: viewing protein secondary structure predictions within large multiple sequence alignments. Bioinformatics, (2017); 33(24):3991–3992
- Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC bioinformatics, (2010); 11(1):1-10
- Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics, (2009); 25(21):2744-2750.
- Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Research, (2010); 38:W214-W220.
- Pathak RK, Lim B, Park Y, Kim JM. Unraveling structural and conformational dynamics of DGAT1 missense nsSNPs in dairy cattle. Scientific Reports, (2022); 12(1):4873.
- Chai CY, Maran S, Thew HY, Tan YC, Rahman NM, Cheng WH, et al. Predicting Deleterious Non-Synonymous Single Nucleotide Polymorphisms (nsSNPs) of HRAS Gene and In Silico Evaluation of Their Structural and Functional Consequences towards Diagnosis and Prognosis of Cancer. Biology, (2022); 11(11):1604.
- Zhang M, Huang C, Wang Z, Lv H, Li X. In silico analysis of non-synonymous single nucleotide polymorphisms (nsSNPs) in the human GJA3 gene associated with congenital cataract. BMC molecular and cell biology, (2020); 21(1):1-3.
- Tastan O, Klein-Seetharaman J, Meirovitch H. The effect of loops on the structural organization of α-helical membrane proteins. Biophysical journal, (2009); 96(6):2299-2312.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0