

Review Article
Annotation and curation of hypothetical proteins: prioritizing targets for experimental study
Muhammad Naveed1,*, Zoma Chaudhry2, Zeeshan Ali2, Mahnoor Amjad2, Fizza Zulfiqar2, Ali Numan2
Adv. life sci., vol. 5, no. 3, pp. 73-87, May 2018
*– Corresponding Author: Muhammad Naveed (Email: naveed.quaidian@gmail.com)
Authors' Affiliations
2- Department of Biochemistry and Biotechnology, University of Gujrat, Gujrat, Pakistan
Abstract![]()
Introduction
Methods
Discussion
Conclusion
References
Abstract
Completely sequenced organisms have some uncharacterized proteins that are gene-encoded products. These proteins can be predicted through in-silico approaches and their biological activities are not proved by experimental evidence and known as hypothetical proteins (HPs). These proteins are important due to their excessive involvement in different cellular and signaling pathways. Structural and functional characterization of HPs reveal crucial roles in microorganisms, especially in pathogens related to human diseases. Here, we discussed all possibilities of in-silico analysis tools and other recently reported methods for hypothetical protein characterization and biomedical applications, including drug and vaccine development. Different methodologies, including meta-proteomics have been used to study protein expression by identification of HPs and comparative genomics have also come under observation due to the emergence of evolutionary study among different organisms. Structural characterization of proteins acts as a base for their functional prediction, novel drug target identification for disease treatment, vaccine production and sero-diagnosis. HPs have played major roles in different vital phenomenon for life including host adaptation, wound healing and chemotaxis. In the current era of drug and antibiotic resistance, HPs can be novel targets to treat related diseases. Identification and characterization of most HPs are under observation and will be the most promising genomic and bioinformatics techniques in structure-based drug designing and vaccine production in future.
Keywords: HPs, structure-based drug design, vaccine development, bioinformatics, function annotation
Introduction![]()
Proteins are diverse biomolecules which are involved in almost all the biological processes of an organism. They provide both specific structural and functional analogy to an individual. The knowledge of proteins has been accomplished via biochemical and genetic experiments [1]. Classification of proteins has been done based on their structure, functions, physiochemical properties and especially on their contribution to metabolic pathways. Current advances in biological techniques have led to the rapid sequencing of microbial genomes and multicellular organisms, paving the way in rapid characterization of genes and their protein products [1,2]. But there remains many uncharacterized genes and their products known as hypothetical proteins (HPs) [3].
A hypothetical protein is defined as the gene or its product that is predicted to be expressed from open read frame (ORF), but there exists no evidence of translation from that ORF experimentally. The existence of such proteins can be merely predicted by computational tools [4,5]. Some proteins are either analogous to proteins having unknown function, so referred as “Conserved Hypothetical” proteins. The genomic projects retain several proteins with unknown functions. The ratio is about up to 30 – 40% of the unknown proteins in prokaryotes and countless in animals and plants. It has become essential to characterize HPs through different approaches which can provide evidence of these proteins performing indispensable roles in microorganisms and multicellular organisms [6].
Our current knowledge on gene functions lags behind the DNA sequencing of organisms. As more and more organisms are being sequenced, the burden of assigning functions to genes is increasing. DNA sequencing of an organism is more important when genes of an organism are assigned to their functions. About 30% of genes in newly sequenced microorganisms are HPs. Many efforts are underway to characterize and annotate function of HPs [7]. For these reasons, along with indispensable roles that HPs perform in organisms, their characterization has got much attention. They perform crucial roles in resistance development, host adaptation, induction of chemotaxis and treatment in wound healing [8,9].
HPs are also involved in pathogens triggered disease development [10,11]. Characterization of HPs is important due to their potential applications in disease treatments. Molecular weight, size and function of HPs must be known not only for identification but also for drug design and vaccine development. Information about disease mechanisms is essential for disease treatment due to the involvement of different HPs. Characterization of HPs is performed by using different advanced methods, bioinformatics, proteomics and genomics-based approaches are the most powerful tools in prediction and characterization of HPs in genomes.
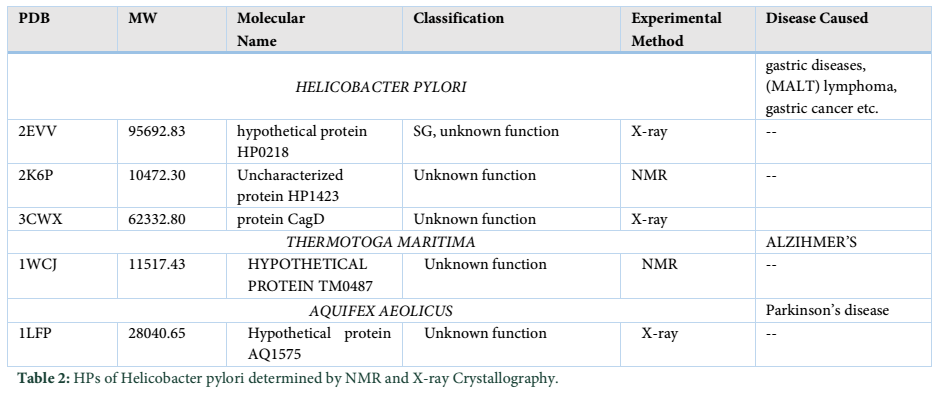
Along with immuno-proteomics, these approaches are effective in prediction and identification of HPs [12]. The immune-proteomics also provides a platform for diagnosis assay and vaccine development. After in-silico analysis of HPs, biochemical characterization is necessary, and it can be carried out through different chromatographic techniques experimentally [1]. Most HPs have the potential applications in drug design, elucidation of disease mechanisms and pathways, where structures and functions are clearly identified. Structural prediction of HPs is carried out by X-ray crystallography and nuclear magnetic resonance (NMR), while in-silico approaches use various online software and databases. For precise and accurate structure analysis, X-ray crystallography patterns and NMR spectroscopy are used frequently [13].
Function prediction is an ultimate goal for almost all uncharacterized proteins. It is important to assign functions to uncharacterized ORFs, and in understanding the roles of HPs in metabolic and disease pathways. For functional prediction, different homology techniques are used, where function is annotated based on gene family or conserved regions, species evolutionary relationship. Two such methods are discussed here are: Clustral approaches and comparative genomics. After characterization, computational verification is done by using bioinformatics tools. HPs are being explored for biomedical applications; in understanding disease mechanisms, drug design and vaccination. Microbial life is being explored to identify potential targets in the era of antibiotic resistance. As pathogens show resistance against many antibiotics available, so new potential targets are needed to overcome pathogen resistance.
In the previous study, structural and functional annotation of different HPs was carried out by means of several experimental and bioinformatics-based approaches. The aim of this review is to characterize the HPs, using different bioinformatics and biochemical approaches to ensure their extensive roles in microorganisms. It also addresses that how this characterization has been proved helpful in correlating HPs with different diseases and their treatment. After complete characterization, role of HPs as drug target and structure development for drug designing come under focus due to requisite applications in disease treatment.
Methods![]()
Literature survey and selection criteria
A well-ordered search was conducted by operating Google scholar and science direct web browser, providing keywords “databases and tools for hypothetical proteins characterization”, “Role of hypothetical proteins”, “hypothetical proteins and experimental techniques”, “tools for structural and functional annotation of hypothetical proteins”, “applications of hypothetical proteins” etc. The literature is then analyzed and screened further according to peculiar contents. In this study, 68 research articles were selected.
Discussion![]()
Characterization of HPs
HPs are uncharacterized gene or gene products and have no significant homology or similarity with any characterized genes or gene products. These genes are predicted by sequencing programs for example, GLIMMER is a sequencing program that finds >97% of genes annotated in literature, but a significant portion of genome have genes that are predicted by software with no homology available in the online databases e.g. NCBI with BLAST analysis. Due to no homology, these genes are considered unique and may catalyze unique functions [5]. During past a few decades, characterization of such proteins is under focus for disease treatment by using experimental and bioinformatics tools [14]. For example, the Helicobacter pylori genome sequence has been commenced in 1997 with 26,695 strains of it. It was shown that its circular chromosome poses 1,590 ORFs that are protein coding regions. From this analysis, it was thought that there is minute region of ORF whose function is unknown. After the long term analysis of its 36 sub-species, it was known that, out of 60,000 genes and translated region, 40% (23,161) are functionally unidentified [15].
Proteomics based identification of HPs expression
From the beginning, there were no evidences for genes expression related to HPs. So, the first step in hypothetical protein characterization is to investigate the expression of genes also termed as hypothetical. Among many methods of HP expression investigation, such as mass spectrometry, proteomics approaches including bottom-up and short-gun approaches. Proteomics and mass spectrometry helps in identification of HPs being expressed in an organism. Accurate Mass and Time (AMT) tag MS proteomics methodology is a proteomic approach based on mass spectrometry [16]. It is relatively easy as it has peptide database and uses mass spectrometry for HP identification [5,17].
A study conducted by [5] for protein investigation of S. oneidensis reveals that MR-1, Mass and time (AMT) tag MS proteomics methodology has been used not only for identification of HPs expression but also their cellular locations by recognizing their peptide signals. Protein lysate is subjected to trypsin for protein digestion which is subjected to MS. After MS coupled chromatography analysis, series of software are applied to identify peptides that are expressed by hypothetical genes. On the other hand, bottom-up is the approach for protein analysis at the level of peptides, known as short-gun proteomics. In shot gun method, the mixture of peptides is fractionated and subjected to LC-MS analysis. Identification of peptides takes place by comparing the velocipede mass spectra obtained from fragmentation along with mass spectra generated from in-silico digestion posture of a protein database [17].
Meta-proteomics not only gives identification but also characterization of the HPs via short-gun method. Recently, it has revealed gene composition in the gut of micro biota for human gut system. But information is still insufficient about gene types, functions and expression. For it, number of protein and peptide-based databases are available for prediction analysis via short gun method. HPs are identified by comparing with the reference databases. About 80% of HPs show similarity with reference sequences present in database and hence the function is predicted by comparison [18].
Biochemical characterization
After identification of HPs, biochemical characterization is another basic and major step in HPs characterization. As it provides information about amino acid sequence, protein molecular weight, physiochemical properties and stability, and protein function. Km, Vmax, and Kcat values are also determined by biochemical analysis of proteins. For biochemical characterization, it is important to separate proteins from the mixture. It was done by different chromatography techniques i.e. affinity or ion exchange chromatography or gel electrophoresis can be additionally utilized for this purpose. Identification of protein expression by proteomics and MS gives result in form of peptides. These peptides are then used for biochemical activities [7,19]. Biochemical characterization can also be done by using cloning and expression of recombinant genes in vectors followed by protein purification and biochemical testation procedures.
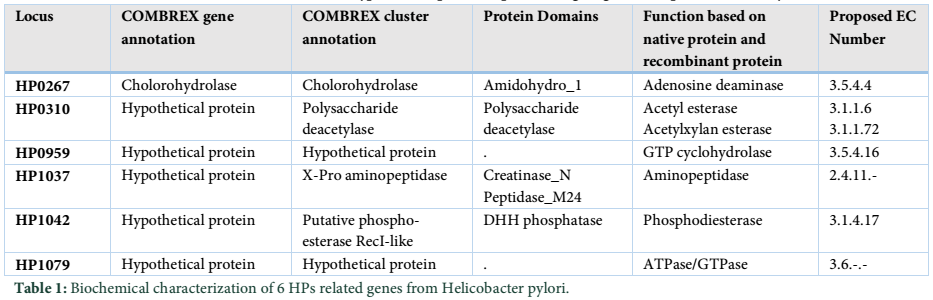
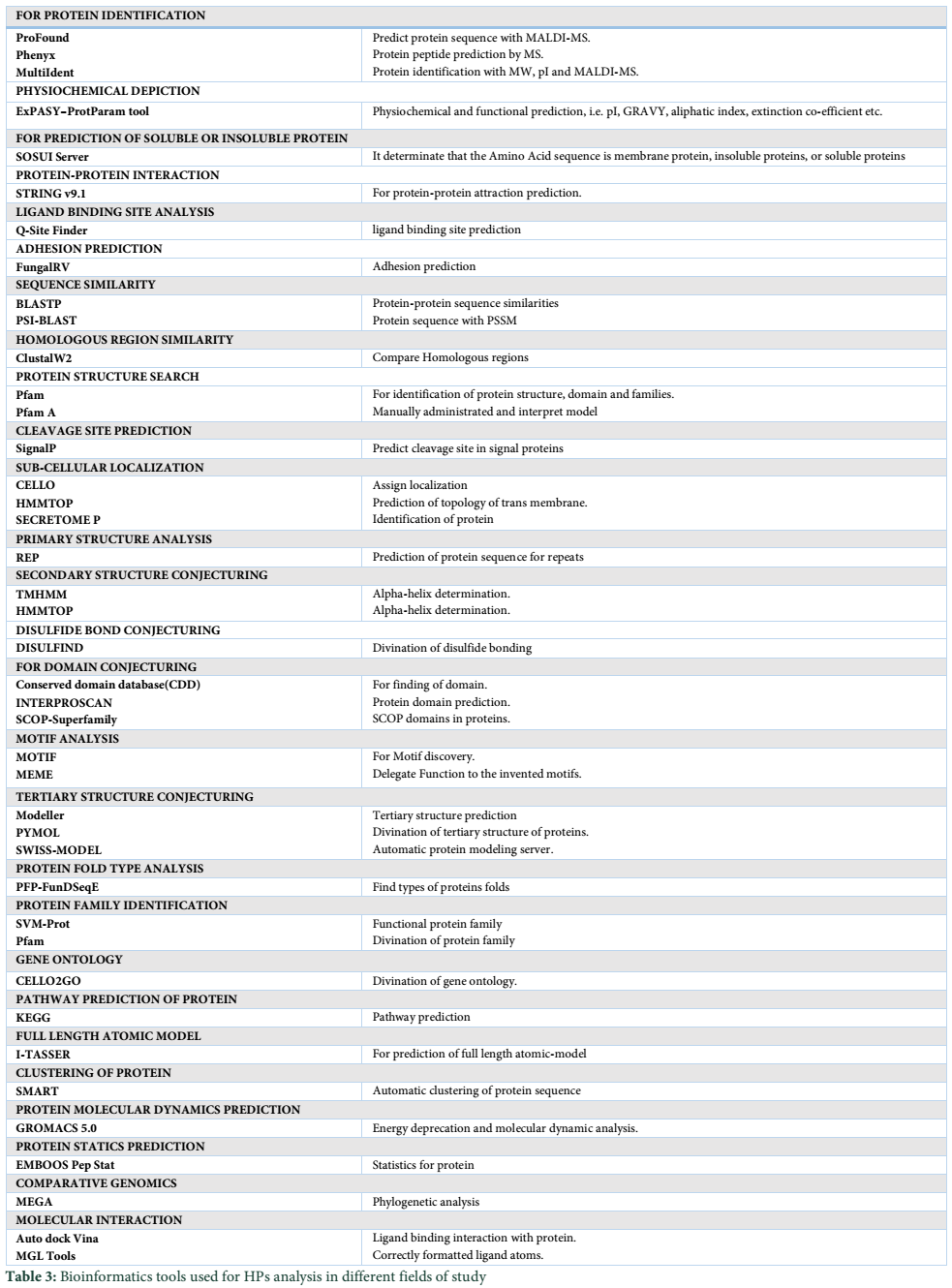
Biochemical characterization is done to annotate general functions of proteins (proteases, phosphates, kinases etc.). Biochemical characterization of [7] H. pylori HPs was tested by gold nanoparticles bounded substrate. Different HPs were subjected to attach their respective substrate. In this way, the general function of HPs is determined (Table 1) but now most of the biochemical and biophysical characterization are performed by different bioinformatics-based tools (Table 3).
Structural prediction of HPs
Structural analysis of a protein is important for studying its different parameters. For example: conformational changes, rotation of bond angles along the axis, binding to its target and protein activity [20]. Like biochemical characterization, structural and functional prediction of proteins also includes experimental and in-silico approaches [21,22]. Two familiar experimental approaches are involved in the structural determination of HPs: X-ray crystallography and NMR [23]. X-ray crystallography determines structure on basis of diffraction pattern. Pre-requisite for this technique is to crystallize protein by addition of solvent (2-metyl, 2,4 Penta-diol: an organic salt) after its purification [20].
NMR spectroscopy has been used in determination of protein folding and protein dynamics. This technique utilizes the magnetic properties of atomic nuclei. For protein structural determination, the N and C termini are labeled and are resolute by different spectrum such as NOESY/COSY, utilized in relation with ATNOS/CANDID/DYANA software for the tenacity of 3D structural analysis of HPs. Knowing that, 3D structural analysis of hypothetical protein by X-RAY and NMR is difficult to perform and time-consuming with the advantage of accuracy and precision [23].
After successful experimental analysis, it is easy to know the structural and functional relationship of protein by comparing its structure to different structures present in a database. This method uses different software that are based on certain bioinformatics algorithms [20]. Alone bioinformatics can also give the structural prediction of protein on basis of high sequence identity and other parameters. By comparing the sequences, one can deduce protein structure, its evolutionary relationship and its respective family [24].
Functional prediction of HPs
The ultimate goal of a hypothetical protein characterization is function annotation or assigning a function to gene. Function prediction has got importance in pathogens for medical prospective. For example, genome of Mycobacterium tuberculosis contains 50% unknown proteins with uncharacterized structure and function. It has pro-glu and pro-pro-glu proteins family which are HPs [26]. Functional prediction of these HPs is very important. It helps to understand more precisely about disease causing pathogens and help scientists to design drugs and antibiotics more accurately. Functional prediction also includes the study of drug resistance, improved biosynthetic pathways as well as targeted antibiotics [27]. Bioinformatics have revolutionized the functional annotation of HPs. Bioinformatics tools based on certain algorithms use different strategies to predict protein function, their sub-cellular locations and physiochemical properties.
Here we discuss, function annotation based on 3D structure, Clustral approach, comparative genomics and different tools used in bioinformatics for functional prediction of HPs [28,29]. 3D structure of HPs can help in function prediction of such proteins. During the process of evolution, folding patterns remain conserved. These conserved patterns are hints to predict functions. Structure based comparison is even helpful when sequence-based comparisons are futile. Structure based comparisons find homology between proteins of known function and HPs. For example, an HP named, MJ0577 from Methanococcus jannaschii contains a 1.7Ao ATP binding domain, predicting that MJ0577 is an ATPase. Other HPs of this family also contain same motif or ATP binding site. So, it was concluded that a same function is performed by all members of this family [25].
Clustal approaches
Clustal approach is used for the functional annotation of HPs present in prokaryotes. Prokaryotic organisms have usually single chromosome and most of the genes are polycistronic, found in clusters. Each cluster contains genes for different proteins. This approach is based on fact that genes of a single cluster or Run are involved in similar functions. Run is a cluster of genes in prokaryotic chromosome, that occur in same strand and gap between these genes in a cluster is about 300bp, and a pair of genes in a ‘run’ is known as ‘close’. It means that any gene from a cluster has the similar function to others genes of that cluster [29]. This approach has been used in function annotation of E. coli hypothetical protein. It belongs to cluster having genes: pgm (2,3- bis, phosphoglycerate independent phosphoglycerate mutase, EC 5.4.2.1), pgk (phosphoglycerate kinase, EC 2.7.2.3, gap (glyceraldehyde 3- phosphate dehydrogenase, EC 1.2.1.12), tpi (triose phosphate isomerase, EC 5.3.1.1) and eno (enolase, EC 4.2.1.1). All these genes are involved in the transcription regulation functional annotation to HP [30]. This hypothetical protein shows homology to the hypothetical transcriptional regulator of Bacillus megaterium predicting correct functional identification [21]. Evolutionary conserved genes are successful in prediction of protein function. Orthologous genes retain their function in different species in course of evolution due to the common ancestor. Orthologous co-expression is a way to predict the function of conserved proteins [31]. Clustral approach is used to build database for clusters of orthologous group (COG), as the coupling of function to members of genes in a cluster [29]. Orthologous co-expression is related to protein-protein interaction (PPI), where two different organisms are studied on basis of protein synthesis, came from a common ancestor (Figure 1) [30].
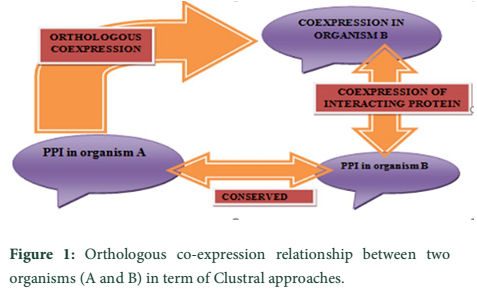
Comparative genomic
Conserved HPs are found in organisms of different phylogenetic lineage [8]. Comparative genomics is the study of genomics to evaluate the relationships between genomes of different species. The concept of comparative genomics is based on assumption that a group of proteins that function together is also evolved or eliminated together, during evolution. So, evolution linked proteins are either eliminated or preserved in next species. Proteins which show homology in different species are functionally linked [29]. Different fluctuations in environment may have the effect on each protein, produced in the body. Thus, reducing the efficiency of natural selection increases fix presumption of all mutations, including that are strongly deleterious on verge [32]. Mutation in HP may have a negative effect, causing diseases in the body.
To know the phylogenetic linkage, we create a phylogenetic profile of organisms that contains homologs proteins. These profiles are used to get phylogenetic linkage between different proteins. Those with similar profiles are functionally linked without knowing the amino acids sequence of proteins. So, without amino acid sequence, the function of HPs can be annotated by this method [29,33]. For example, a mutation of tc0668 gene of hypothetical protein in chlamydia muridarum; a murine model of human urogenital C. trachomatis having severe disease in the upper genital tract of female mice. The mutant contains a TC0237Q117E mutation which increases in-vitro infection. The genetic assay reveals that a nonsense mutation in G216* of about 408 amino acids does not produce an observable product. In the absence of Tc0668, intracellular growth and infection caused by C. muridarum are unaffected. Thus, TC0668 is demonstrated as exigent chromosome that encodes urogenital bug factor of C. muridarum [34]. A tool used to create the phylogenetic tree is MEGA (Molecular Evolutionary Genetics Analysis). It uses multiple features i.e. aligning sequences, estimating evolutionary distance, build phylogenetic tree, marking gene domains and compute sequence statistics to create phylogenetic Tree [35].
Studies of auxotrophic mutants claims that conserved HPs are essential for survival [29]. X-Ray and NMR spectroscopy studies reveal the function of HPs that causes mutation in an individual. On the basis of it there are two types of HPs: unknown-unknown and known-unknown. The former is that in which prediction of biochemical activity is not known while later means the prediction of biochemical identification takes place [8,34].
Bioinformatics-based analysis
Bioinformatics-based tools and algorithms are replacing most of the wet lab experiments in predicting functions of HPs. Bioinformatics tools process function prediction starting from sequence retrieved from NCBI or UniProt database, physicochemical characterization of proteins, finding subcellular locations and functional prediction (Table 3) [28,36,37].
Computational verification
After successful structural and functional analysis of HP, it is necessary to verify the structure and function. ERRAT, Procheck and Ramachandran plot are basic bioinformatics tools for computational verification of 3D predicted structures of proteins. ERRAT is an algorithm for protein structure verification used to evaluate the structure obtained from crystallographic technique [38]. On basis of stereo-chemical quality, Procheck analyzes protein structure by its geometric analysis and produces a number of plots that give an overall assessment of the structure quality as compared to well-refined structures of the same resolution. It also highlights those regions that may need further investigation [39].
Ramachandran plot uses computational models of small polypeptides with variable psi and phi angles to find stable conformations. For stable conformation, an individual structure was analyzed for close association between atoms. So, phi and psi angles that cause collision among atoms correspond to sterically disallowed conformations of the polypeptide backbone, Mostly, it is used for verification of secondary structures e.g. alpha-helix and beta-sheets [40].
Roles of HPs
Characterization of hypothetical protein revealed that HPs play vital roles in different organisms. HPs are involved in specific pathways, regulating different mechanisms in the body. Of these several roles, some most important roles are as follow:
Role in host adaptation
The host adaptation is the capability of the pathogen to troll and infect the host and cause diseases. The absence and presence of HPs show the relationship with host adaptation. For example, assembly of Mycoplasma depends on pan domainome which is not correlated with the host cell. Pan domain is an apple like domain having functional utility among the protein-protein interactions. The collation between pan and core domainome of mycoplasma with the minimal synthetic organism. JCVI-Sync3.0 evaluate it in order to determine the role of hypothetical protein in host adaptation as well as synthetic minimal life. A synthetic organism is ''artificial organism'' that can reproduce, exist and maintain itself having properties like living but they can't imitate life so also called ''minimal cells'' [41]. By comparing the domainome of mycoplasma with JCVI-Syn3, it is commenced that all domains that are present in mycoplasma also exist in a minimal synthetic organism and show host specific relationship [42].
Role as multiplex
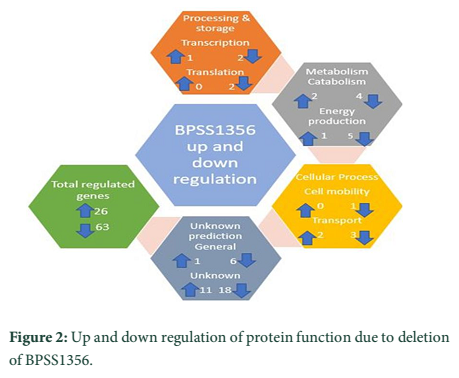
Single HPs have conserved functional domains that provide a prediction about the shared part and give division with other possible domains that are not contributed [15]. There are number of single HPs that have multiple roles, i.e. BPSS1356 can up and down regulate at the same time (Figure 2).
For instance, Burkholderia is a pathogen that causes melioidosis. It can live and survive within the host cell. A pull-down assay determines the presence of HPs that reveals the presence of bound hypothetical protein i.e. BPSS1356. The deletion of this protein performs multiple function i.e. biofilm formation and lower cell growth. Electron microscope analysis of mutant cells shows that the shrinkage of cytoplasm leading to condition plasmolysis Membrane transporters is down regulated by the protein variation. The expropriation of this gene also down regulated the transcription of genes for glycerol metabolism, type 3 secretion system and arginine diaminase. It is, therefore, BPSS1356 has vital multiple regulatory roles [43].
Resistance development
Hypothetical protein plays a dominant role in resistance development i.e. hypothetical protein in Cercospora nicotianae developed resistance against their own toxins. Cercosporin induce universal toxicity to the cell in Cercospora. It is photo activated toxin that develops fungus i.e. Cercospora a strong pathogen against host cell i.e. plant cells. The toxicity is due to reactive oxygen species formulation. Quantitative analysis of PCR tells that there are 20 genes that encode HPs and only two of these, 24cF and 71cR, provide resistance against toxicity of Cercosporin. Expression and transformation of 24cF and 71cR propose that 71cR provides crucial and increased resistance to Cercosporin toxins as compare to 24cF [44].
Role of membrane HPs
There are many genes that encode HPs which perform the vital function in the membrane. For example, Legionella pneumophila (Lp) is a water born pathogen which expresses the gene Ipg1659 in water. Deletion of this gene affects the membrane integrity, tolerance capacity and cell morphology. Due to its role Lpg1659 was given the name LasM of Legionella aquatic survival membrane proteins. HPs act as a transporter of metal ions and maintain integrity.
Role in the induction of chemotaxis
The fundamental property of living cells is to respond to the change in the environment [46]. Extracellular ligands gradients are detected by cells and then lead them to move either towards the source (chemoattractant) or away from the source (chemo repellent).It is known as chemotaxis [47]. HPs are essential for the production of chemotaxis. For example, Spirochete Borrelia burgdorferi has five chemotaxis proteins known as MCPs (methyl-accepting chemotaxis proteins). Out of five, one is BB0569 which is a HP with conserved domains.
The mutation in BB0569 results in movement in one direction and failure to respond against chemoattractant. The BB0569 present at the cell poles and is essential for clustering of chemoreceptors at cellular poles. Hence, it is indicated that BB0569 has a vital role in chemotaxis that is a distinct feature of spirochetes [48].
Role in wound healing
The care and treatment of skin wounds is a major issue due to health expenditures as skin wounds become severe specially in case of clotting in dermal blood vessels, inducing acute hypoxic condition [49]. It is the major factor of stress that leads to remodeling of basal keratinocytes to initiate epithelization. The lateral migrated keratinocytes secreted an extracellular protein known as heat shock protein 90 alpha. This is one of the HPs involved in wound healing as they engaged low density of lipoprotein receptors related protein-1 (LRP-1) cell receptors and also work as the autocrine factor in order to stimulate the keratinocyte migration that is involved in re-epithelialization. The other factor is paracrine which builds the fibroplasia, i.e., the stimulation of the exodus of dermal fibroblast, neo vascularization i.e. the microvascular endothelial cell construction. Hypoxia activate extracellular protein (hsp90a) which act as a major regulator of skin wound repairing [49].
Presence of HPs in pathogens
HPs are present almost in every organism even in model organisms like E. coli, multicellular organisms and viruses. Their presence in pathogens, and other microorganisms have created great interest in biomedical applications. These proteins are involved in major metabolic and disease pathways of pathogens related to both animals and humans. Here, we discuss some pathogens with their HPs [14]. Helicobacter pylori and Vaccina virus cause the gastric ulcer and smallpox respectively in humans with respect to their HPs. H. pylori genome sequence has been commenced in 1997 with 26,695 strains of H. pylori. It was shown that its chromosome is circular, with 38.8% GC content and 1,590 open reading frames that are protein coding regions. From this analysis, it was thought that there is a minute region of ORF whose function is unknown. After the long term analysis of 36 sub-species of this organism, it was concluded that out of 60,000 genes and translated region, 40%(23161) of the genes function are unidentified ,which are translated to hypothetical protein [15].
Similarly, Vaccina virus is a distinctive virus and belongs to the class of poxvirus, its genome size ranges from 130 to 300Kb containing linear and dsDNA. The genome of vaccina virus encodes about more than 200 proteins in which 64% are identified or their function is known while 36 % of the protein function are unidentified and that are termed as HPs [50]. Recently, a hypothetical protein responsible for Fanconi anemia is FANCF1 is discovered which is an analogue to BRCA2 [48]. Aedes aegypti is responsible for the out brake of many diseases caused by HPs present in the saliva of A. aegypti. In recent studies, three proteins are identified with molecular weight of 77, 58, 54, and 37kDa for vaccine production, respectively [51].
C. trachmatis is responsible for causing trachoma and sexual transmitted disease “Chlamydia” in human, contains 887 genes for protein translation. Of these, 269 are regarded as HPs, that include 24 proteins responsible for the disease. For example, 084134 encodes for serum resistance.084024, 084052, 084054, 084103, 084107, 084177, 084184, 084226, 084349, 084356, 084363, 084364, 084365, 084472, 084480, 084534, 084556, 084582, 084593 084654, 084671, 084700, 084721, 084749 are determined by VICMPred tool as virulence proteins [52]. Salmonella enteritis causes typhoid fever and illness by utilizing its hypothetical protein:HPR-27 [22].
Parkinson’s disease is caused due to the accumulation of Alpha-Synulein as it is drawn from the ubiquitin degradation pathway. Recent studies have shown that hypothetical protein HP-CAB-55973 processes ubiquitin-like motifs. Some HPs responsible for this are known including (Q9BTE6) CGI-83 protein, (Q9Y392) CG11334 and (Q9BV20) [53].
HPs and drug development
Drug development is a complex process which requires potent targets having high efficacy for drug molecule [54]. Many strategies for designing a drug are known. But in recent years, structure-based drug designing is one of the most convenient methods, which includes the design of ligand considering the structure of the target. The science of structure based drug design is influenced and enriched by proteomics, genomic sequences and bioinformatics, making it a promising field in drug discovery and development [55]. Structure-based drug design started in mid of 80s’ and now has become most powerful tool in drug discovery [56]. A drug target can be a protein, DNA, RNA or receptors present on the surface of cell. Among these protein targets, HPs can be the novel targets. Target should only be present in the pathogen, and not in the host, so that ligand could not block the normal pathways of the host.
In the current era of drug and antibiotic resistance, when most of the pathogens are developing resistance, novel drug targets are required [57]. Along with these for treatment of many other diseases new targets are to be searched. Bioinformatics and genomic sequencing are major approaches in novel drug target researches. Uncharacterized ORFs including HPs are being explored by these approaches for drug development [58]. Functional annotation of HPs has given bulk information about the pathogenicity of the invader and metabolic pathways of a pathogen that can be blocked by novel drugs [52, 59, 60]. In drug development against infectious diseases especially antibiotic resistance developed bacteria, major concern is proteins involved in disease mechanism directly, and HPs can be major targets as they perform vital roles in pathogens including disease mechanisms called virulence factors. Virulence factors are involved in causing virulence by pathogens [60]. Direct involvement of these proteins in disease mechanism make them a good target to control disease effectively and instantly.
The process of drug development for HPs starts with sequence retrieval and characterization of proteins structurally and functionally by using different bioinformatics tools (Table 3). Comparisons between HP sequences of the pathogen, model organism and host are done by using different softwares. These comparisons give information about conserved regions of proteins in host and pathogen followed by homology comparison. Proteins having little homology with the host are selected for further molecular docking analysis.
Docking is the process through which interaction between two compounds, ligand and target are predicted by using computer algorithms. From docking score best target is selected which has well defined pocket. Docking screens for ligands in database and score according to interaction with a target. Ligand with the best score is tested using high through put screening in lab, if results are positive further modifications are done to ligand and pre-clinical trials are conducted [59].
For example, eukaryotic pathogens of genus Leishmania, cause diseases in humans. These parasites are limited by many factors and available drugs have many side effects including fever, myalgia, arthralgia and liver toxicity. In recent studies, four cytoplasmic HPs were selected for structural and homology comparison to host and docking analysis was performed to select best for drug development. One of these proteins was selected as the potential that has ligand binding capacity and also not present in humans [59]. Recently, a research was made in the characterization of HPs of adenovirus for possible drug targeting and STEAP2 was identified as therapeutic gene target for prostate cancer treatment.
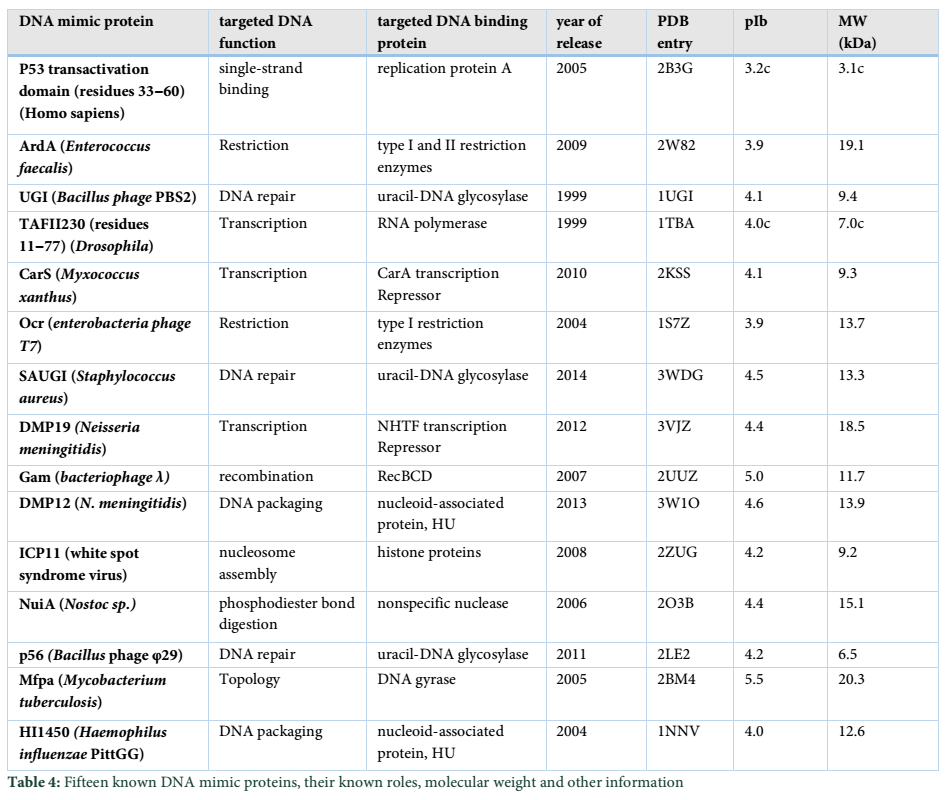
DNA mimic; revolutionised DNA binding proteins
DNA mimic has unique factors to control the DNA binding proteins activity by occupying its own DNA binding sites. The sequences of these proteins are very diverse that make it very difficult to find by bioinformatics search. Only some of these proteins have been reported in these days and fewer of these DNA mimic proteins have been analyzed on the basis of their functions (Table 4). DNA mimics resembles to HPs in having same conserved domains [61]. Like in Neisseria, DMP12 is a DNA mimic protein as its monomer shows the interaction with the dimeric of histone like proteins HU. Moreover, here is the NMB2123 is hypothetical protein which acts as the mimic proteins i.e. DMP12.
Through gel filtration and analytical ultracentrifugation, it was found that DMP is present in its monomeric form when it interacts with bacterial histone like HU-proteins. Their crystalline structure was also studied and with help of isothermal titration calorimeter, the binding affinity between DMP and Messenia has been determined. Thus, the interaction between DMP12 and Hu-protein considered as an instrument for the stability of nucleoid in Neisseria [62].
DNA mimic proteins are able to copy a specific charge distribution with the two negatively charged amino acids: glutamic acid (E) and aspartic acid (D). While the Known DNA mimic Proteins having different shapes, structures and different negative charge distribution on their Surface [61]. Being so important like DNA molecules, DNA mimics perform various functions in body including: 1) DNA repair 2) DNA packaging 3) Topology 4) phosphodiester bond digestion 5) Nucleosome Assembly 6) Transcription 7) Histone protein 8) Recombination 9) Restriction 10) Single-strand binding [61].
Applications of HPs
HPs found its major applications in the field of clinical, medicine, pharmaceuticals and molecular biology due to extensive study related to bioinformatics. In the present era, due to bioinformatics, these proteins have been proved helpful in the development of different new strategies and vaccines to diagnose and treat diseases [63]. Important applications related to HPs are as follow:
Novel drug development against resistant pathogens
Drug resistance is a major problem in infectious disease treatment and new drug targets are to be identified in resistant pathogens. Expression proteomics and bioinformatics tools have emerged for the study of drug resistance. These studies of proteomics and bioinformatics have proposed HPs as novel targets. Using bioinformatics tools; like molecular docking, protein-protein analysis; HPs are characterized by drug binding in silico models [64, 65].
Vaccine production
HPs are serving in vaccine production against pathogens. By using bioinformatics tools conserved domains are identified, that may predict the function of proteins. The potential proteins are subjected to physiochemical analysis, sub cellular location analysis, protein-protein interaction analysis. The next step is to identify the epitope; by in silico method; that can recruit T and B cells of the host, their functions and subcellular locations. Highest scored HPs are then used in vaccine production [66].
Marker for disease identification
Like HPs, expression of many proteins is up or down regulated in different disease. As their function is unknown, so their changing expression may be useful for their functional determination in a specific organ. For example, a HP of 28.5kDa is significantly upregulated in the fetus with Down’s syndrome. HPs may act as markers for disease or their function can be predicted in normal and disease mechanism [67].
Serodiagnosis; an evaluation for pathogenic diseases
Serodiagnosis of proteins is an important phenomenon to evaluate pathogenic diseases. Mostly, it gives poor results in sensitivity and selectivity for unknown proteins. Here, bioinformatics tools are the alternatives to avoid this problem. For example, Tegumentary leishmaniosis is endemic in Latin America, caused by pathogen Leishmania Brasiliense’s: a eukaryotic parasite [68]. The present serodiagnosis of parasite is less sensitive and specific, making major problem in early diagnosis of disease. During past few years, enzyme-linked immunosorbent assay (ELISA) is used to evaluate performance of pathogen by its hypothetical protein LbHyM with poor sensitivity and selectivity. Recently, prediction of LbHyM was done by using bioinformatics tools including sequence retrieval, physicochemical characterization and molecular interaction with the host cell to cause infection [63].
Conclusion![]()
Research is underway to understand HPs, their structural and functional annotation as they are important in metabolic pathways and many diseases. HPs are being characterized, are paving the way in a better understanding of microbial metabolic pathways, drug development against pathogens, and disease control strategies. Structural and computational based drug development has revolutionized the process with new targets. DNA mimic being resemblance to HP performs very essential functions and their presence in binding pockets have a crucial role at molecular level due to charge specificity. Due to its major concern nowadays, the proportion of HP is increasing in NCBI. Recent advances and efforts to understand HP will bring revolution in the biological system. In future, HPs will find many other applications that will revolutionize the disease treatment, diagnosis, vaccine production in resistance developed pathogens and in development of new fields of sciences.
Acknowlegdement
The authors appreciate the technical assistance offered by the Joeman Adaeze, Boutiti Promise and Cosmos Obi of the Department of Pharmacology and Toxicology, Niger Delta University Amassoma.
Conflict of Interest Statement
The authors declare that there is no conflict of interest regarding the publication of this paper.
References![]()
- Mertens HD, Svergun DI. Combining NMR and small angle X-ray scattering for the study of biomolecular structure and dynamics. Archives of biochemistry and biophysics, (2017); 628: 33-41.
- Jacobs T, Williams B, Williams T, Xu X, Eletsky A, et al. Design of structurally distinct proteins using strategies inspired by evolution. Science, (2016); 352(6286): 687-690.
- Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proceedings of the National Academy of Sciences, (1999); 96(8): 4285-4288.
- Enany S. Structural and functional analysis of hypothetical and conserved proteins of Clostridium tetani. Journal of infection and public health, (2014); 7(4): 296-307.
- Elias DA, Monroe ME, Marshall MJ, Romine MF, Belieav AS, et al. Global detection and characterization of hypothetical proteins in Shewanella oneidensis MR‐1 using LC‐MS based proteomics. Proteomics, (2005); 5(12): 3120-3130.
- Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, et al. mapman: a user‐driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. The Plant Journal, (2004); 37(6): 914-939.
- Choi H-P, Juarez S, Ciordia S, Fernandez M, Bargiela R, et al. Biochemical characterization of hypothetical proteins from Helicobacter pylori. PLoS One, (2013); 8(6): e66605.
- Galperin MY, Koonin EV. ‘Conserved hypothetical’proteins: prioritization of targets for experimental study. Nucleic acids research, (2004); 32(18): 5452-5463.
- Shahbaaz M, Ahmad F, Hassan MI. Structure-based functional annotation of putative conserved proteins having lyase activity from Haemophilus influenzae. 3 Biotech, (2015); 5(3): 317.
- Hava DL, Camilli A. Large‐scale identification of serotype 4 Streptococcus pneumoniae virulence factors. Molecular microbiology, (2002); 45(5): 1389-1406.
- Hung M-C, Link W. Protein localization in disease and therapy. J Cell Sci, (2011); 124(20): 3381-3392.
- Yu CS, Chen YC, Lu CH, Hwang JK. Prediction of protein subcellular localization. Proteins: Structure, Function, and Bioinformatics, (2006); 64(3): 643-651.
- Brosch R, Gordon SV, Garnier T, Eiglmeier K, Frigui W, et al. Genome plasticity of BCG and impact on vaccine efficacy. Proceedings of the National Academy of Sciences, (2007); 104(13): 5596-5601.
- Naveed M, Kazmi S, Anwar F, Arshad F, Dar T, et al. Computational Analysis and Polymorphism study of Tumor Suppressor Candidate Gene-3 for Non Syndromic Autosomal Recessive Mental Retardation. Journal of Applied Bioinformatics & Computational Biology, (2016); 5(2).
- Park SJ, Son WS, Lee B-J. Structural analysis of hypothetical proteins from helicobacter pylori: an approach to estimate functions of unknown or hypothetical proteins. International journal of molecular sciences, (2012); 13(6): 7109-7137.
- Smith RD, Anderson GA, Lipton MS, Masselon C, Paša-Tolić L, et al. The use of accurate mass tags for high-throughput microbial proteomics. Omics: a journal of integrative biology, (2002); 6(1): 61-90.
- Zhang Y, Fonslow BR, Shan B, Baek M-C, Yates III JR. Protein analysis by shotgun/bottom-up proteomics. Chemical reviews, (2013); 113(4): 2343-2394.
- Verberkmoes NC, Russell AL, Shah M, Godzik A, Rosenquist M, et al. Shotgun metaproteomics of the human distal gut microbiota. The ISME journal, (2009); 3(2): 179-189.
- Ijaq J, Chandrasekharan M, Poddar R, Bethi N, Sundararajan VS. Annotation and curation of uncharacterized proteins-challenges. Frontiers in genetics, (2015); 6119.
- Bixby C, Mahadevan P. Predicting the Function of Hypothetical Protein PANDA_003700 using Computational Analysis Methods; 2016. The Steering Committee of The World Congress in Computer Science, Computer Engineering and Applied Computing (WorldComp). pp. 130.
- Teh BA, Choi SB, Musa N, Ling FL, Cun STW, et al. Structure to function prediction of hypothetical protein KPN_00953 (Ycbk) from Klebsiella pneumoniae MGH 78578 highlights possible role in cell wall metabolism. BMC structural biology, (2014); 14(1): 7.
- Khan A, Ahmed H, Jahan N, Ali SR, Amin A, et al. An in silico Approach for Structural and Functional Annotation of Salmonella enterica serovar typhimurium Hypothetical Protein R_27. International Journal Bioautomation, (2016); 20(1).
- Almeida MS, Herrmann T, Peti W, Wilson IA, Wüthrich K. NMR structure of the conserved hypothetical protein TM0487 from Thermotoga maritima: implications for 216 homologous DUF59 proteins. Protein science, (2005); 14(11): 2880-2886.
- Madden T. The BLAST sequence analysis tool. (2013).
- Shin DH, Yokota H, Kim R, Kim S-H. Crystal structure of conserved hypothetical protein Aq1575 from Aquifex aeolicus. Proceedings of the National Academy of Sciences, (2002); 99(12): 7980-7985.
- Bashir N, Kounsar F, Mukhopadhyay S, Hasnain SE. Mycobacterium tuberculosis conserved hypothetical protein rRv2626c modulates macrophage effector functions. Immunology, (2010); 130(1): 34-45.
- Bidkar A, Thakur N, Bolshette JD, Gogoi R. In-silico Structural and Functional analysis of Hypothetical proteins of Leptospira Interrogans. Biochem Pharmacol, (2014); 3(136): 2167-0501.1000136.
- Kumar K, Prakash A, Tasleem M, Islam A, Ahmad F, et al. Functional annotation of putative hypothetical proteins from Candida dubliniensis. Gene, (2014); 543(1): 93-100.
- Sivashankari S, Shanmughavel P. Functional annotation of hypothetical proteins–A review. Bioinformation, (2006); 1(8): 335.
- Tirosh I, Barkai N. Computational verification of protein-protein interactions by orthologous co-expression. BMC bioinformatics, (2005); 6(1): 40.
- van Noort V, Snel B, Huynen MA. Predicting gene function by conserved co-expression. TRENDS in Genetics, (2003); 19(5): 238-242.
- Ingram JR, Knockenhauer KE, Markus BM, Mandelbaum J, Ramek A, et al. PNAS Plus Significance Statements. PNAS, (2017): 114(22); 5567-5570.
- Thakare HS, Meshram DB, Jangam CM, Labhasetwar P, Roychoudhary K, et al. Comparative genomics for understanding the structure, function and sub-cellular localization of hypothetical proteins in Thermanerovibrio acidaminovorans DSM 6589 (tai). Computational biology and chemistry, (2016); 61226-228.
- Conrad TA, Gong S, Yang Z, Matulich P, Keck J, et al. The chromosome-encoded hypothetical protein TC0668 is an upper genital tract pathogenicity factor of Chlamydia muridarum. Infection and immunity, (2016); 84(2): 467-479.
- Kumar S, Nei M, Dudley J, Tamura K. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Briefings in bioinformatics, (2008); 9(4): 299-306.
- Ijaq J, Chandrasekharan M, Poddar R, Bethi N, Sundararajan VS. Annotation and curation of uncharacterized proteins-challenges. Frontiers in genetics, (2015); 6: 119.
- Singh G, Sharma D, Singh V, Rani J, Marotta F, et al. In silico functional elucidation of uncharacterized proteins of Chlamydia abortus strain LLG. Future Science OA, (2017); 3(1): 66.
- Satpathy R, Behera R, Guru RK. Homology modelling and molecular dynamics study of plant defensin DM-AMP1. Journal of Biochemical Technology, (2011); 3(4): 309-311.
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of applied crystallography, (1993); 26(2): 283-291.
- Ting D, Wang G, Shapovalov M, Mitra R, Jordan MI, et al. Neighbor-dependent Ramachandran probability distributions of amino acids developed from a hierarchical Dirichlet process model. PLoS computational biology, (2010); 6(4): e1000763.
- Irshad M, Munir H. Structural and Functional Characterization of a Hypothetical protein of Streptococcus Pyrogenes: An In-Silico Approach. Journal of Biochemistry, Biotechnology and Biomaterials, (2017); 1: 54-63.
- Kamminga T, Koehorst JJ, Vermeij P, Slagman SJ, dos Santos VAM, et al. Persistence of Functional Protein Domains in Mycoplasma Species and their Role in Host Specificity and Synthetic Minimal Life. Frontiers in cellular and infection microbiology, (2017); 7: 00031.
- Yam H, Abdul Rahim A, Mohamad S, Mahadi N, Abdul Manaf U. The Multiple Roles of Hypothetical Gene BPSS1356 in Burkholderia. (2014); 9(6): e99218.
- Beseli A, Noar R, Daub ME. Characterization of Cercospora nicotianae Hypothetical Proteins in Cercosporin Resistance. PloS one, (2015); 10(10): e0140676.
- Tong S-M, Chen Y, Ying S-H, Feng M-G. Three DUF1996 Proteins Localize in Vacuoles and Function in Fungal Responses to Multiple Stresses and Metal Ions. Scientific reports, (2016); 6.
- Pandey G, Jain RK. Bacterial chemotaxis toward environmental pollutants: role in bioremediation. Applied and Environmental Microbiology, (2002); 68(12): 5789-5795.
- Ward SG. Do phosphoinositide 3-kinases direct lymphocyte navigation? Trends in immunology, (2004); 25(2): 67-74.
- Zhang K, Liu J, Charon NW, Li C. Hypothetical protein BB0569 is essential for chemotaxis of the Lyme disease spirochete Borrelia burgdorferi. Journal of bacteriology, (2016); 198(4): 664-672.
- Woodley DT, Wysong A, DeClerck B, Chen M, Li W. Keratinocyte migration and a hypothetical new role for extracellular heat shock protein 90 alpha in orchestrating skin wound healing. Advances in wound care, (2015); 4(4): 203-212.
- Mahmood MS, Ashraf NM, Bilal M, Ashraf F, Hussain A, et al. In Silico Structural and Functional Characterization of a Hypothetical Protein of Vaccinia Virus, (2016); 1: 54-63.
- Dhawan R, Kumar M, Mohanty AK, Dey G, Advani J, et al. Mosquito-Borne Diseases and Omics: Salivary Gland Proteome of the Female Aedes aegypti Mosquito. OMICS: A Journal of Integrative Biology, (2017); 21(1): 45-54.
- Naqvi AAT, Rahman S, Zeya F, Kumar K, Choudhary H, et al. Genome analysis of Chlamydia trachomatis for functional characterization of hypothetical proteins to discover novel drug targets. International journal of biological macromolecules, (2017); 96234-240.
- Zarembinski TI, Hung L-W, Mueller-Dieckmann H-J, Kim K-K, Yokota H, et al. Structure-based assignment of the biochemical function of a hypothetical protein: a test case of structural genomics. Proceedings of the National Academy of Sciences, (1998); 95(26): 15189-15193.
- Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, et al. A comprehensive map of molecular drug targets. Nature Reviews Drug Discovery, (2017); 16(1): 19-34.
- Anderson AC. The process of structure-based drug design. Chemistry & biology, (2003); 10(9): 787-797.
- Mountain V. Astex, Structural Genomix, and Syrrx. I can see clearly now: structural biology and drug discovery. Chemistry & biology, (2003); 10(2): 95-98.
- Brown ED, Wright GD. Antibacterial drug discovery in the resistance era. Nature, (2016); 529(7586): 336-343.
- Yoneyama H, Katsumata R. Antibiotic resistance in bacteria and its future for novel antibiotic development. Bioscience, biotechnology, and biochemistry, (2006); 70(5): 1060-1075.
- Chávez-Fumagalli MA, Schneider MS, Lage DP, Machado-de-Ávila RA, Coelho EA. An in silico functional annotation and screening of potential drug targets derived from Leishmania spp. hypothetical proteins identified by immunoproteomics. Experimental Parasitology, (2017); 17666-74.
- Cameron TC, Cooke I, Faou P, Toet H, Piedrafita D, et al. A novel ex vivo immunoproteomic approach characterising Fasciola hepatica tegumental antigens identified using immune antibody from resistant sheep. International Journal for Parasitology, (2017).
- Wang H-C, Ho C-H, Hsu K-C, Yang J-M, Wang AH-J. DNA mimic proteins: functions, structures, and bioinformatic analysis. Biochemistry, (2014); 53(18): 2865-2874.
- Tucker AT, Bobay BG, Banse AV, Olson AL, Soderblom EJ, et al. A DNA mimic: The structure and mechanism of action for the anti-repressor protein AbbA. Journal of molecular biology, (2014); 426(9): 1911-1924.
- Lima MP, Costa LE, Duarte MC, Menezes-Souza D, Salles BCS, et al. Evaluation of a hypothetical protein for serodiagnosis and as a potential marker for post-treatment serological evaluation of tegumentary leishmaniasis patients. Parasitology research, (2017); 116(4): 1197-1206.
- Sharma D, Bisht DM. Tuberculosis Hypothetical Proteins and Proteins of Unknown Function: Hope for Exploring Novel Resistance Mechanisms as well as Future Target of Drug Resistance. Frontiers in microbiology, (2017); 8: 465.
- Gazi MA, Kibria MG, Mahfuz M, Islam MR, Ghosh P, et al. Functional, structural and epitopic prediction of hypothetical proteins of Mycobacterium tuberculosis H37Rv: An in silico approach for prioritizing the targets. Gene, (2016); 591(2): 442-455.
- Duarte MC, Lage DP, Martins VT, Costa LE, Carvalho AMRS, et al. A vaccine composed of a hypothetical protein and the eukaryotic initiation factor 5a from Leishmania braziliensis cross-protection against Leishmania amazonensis infection. Immunobiology, (2017); 222(2): 251-260.
- Engidawork E, Gulesserian T, Fountoulakis M, Lubec G. Expression of hypothetical proteins in human fetal brain: increased expression of hypothetical protein 28.5 kDa in Down syndrome, a clue for its tentative role. Molecular genetics and metabolism, (2003); 78(4): 295-301.
- Alves CF, Alves CF, Figueiredo MM, Souza CC, Machado-Coelho GLL, et al. American tegumentary leishmaniasis: effectiveness of an immunohistochemical protocol for the detection of Leishmania in skin. PLoS One, (2013); 8(5): e63343.
![]()
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0