

Short Communication
Phylogenetic study of 46 Ancient Mitochondrial Human Genomes
Aqsa Khan1, Nasir Ali1*, Wajiha Shafique1, Ghani ur Rahman1, Shaker Khan1, Gohar Rahman1, Bilal Ahmad Mian1, Nazia Akbar1, Habib Ahmad2
Adv. life sci., vol. 6, no. 2, pp. 71-75, February 2019
*– Corresponding Author: Nasir Ali (Email: nociralee@gmail.com)
Authors' Affiliations
2- Islamia College Peshawar, Pakistan
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: In the third era of ancient DNA field, it has endured the mesmerising modifications, which should be revealed. From side to side period, analysis of mitochondrial DNA permits to determine the evolutionary relationship among the species, to expose the terrestrial roots of the entities, to standardise the molecular clocks and to study the demographic pasts.
Methods: In the current study we used bioinformatics tools for prediction of mitochondrial haplogroups and phylogenetic analysis. The ancient complete mitochondrial genomes were retrieved from online resources and were further used for phylogenetic analysis to know the evolutionary position of the ancient populations lived thousands of years ago.
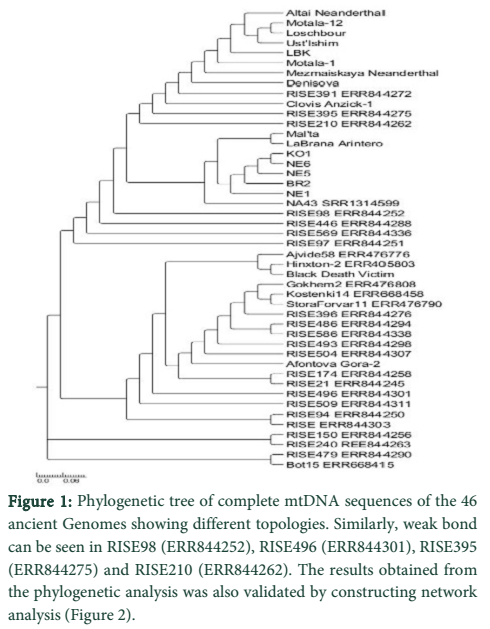
Results: We aligned 46 ancient genomes, collected online and estimated trees by using neighbour-joining, maximum parsimony and maximum-likelihood. Support for nodes was assessed with bootstrap replicates. During our analysis a strong bond between genomes of Altai Neanderthal, Motala 12, Motala 1, Loschbour, Ust'-Ishim, LBK, Mezmaiskaya Neanderthal, Denisova, RISE391(ERR844272), Clovis Anzick-1, RISE395(ERR844275) and RISE210(ERR844262) were found. In this context these ancient samples recommended the presence of a mutual earliest genomic signature.
Conclusion: A significant population immigrations and alternates, accountable for influencing main parts of current demographic structure together in Europe and Asia is supported by the Bot15 (ERR668415) and RISE family. In the initial bronze period, ancestral similarity among these populations also share the theorised blow-out of Indo-European languages. Mechanisms of pathogen development and alteration for evolving and reappearing toxicities is also explained by this study. We aim that this study will help researchers in understanding the evolutionary position of ancient populations resided around the world.
Keywords: Genomes, Mt DNA, Phylogenetic, Ancient DNA
Introduction![]()
Owed to certain of its distinct characteristics for example high copy number per cell, crossing over or lack of recombination, high substitution percentage, and a maternal manner of inheritance, a vital tool to understand human evolution is genetic exploration of mitochondrial DNA [1]. The d-loop that contains not more than 7% of the mtDNA genome is responsible for maximum evolutionary studies that comprised mtDNA arrangements [2]. Interpretations drawn from the d-loop alone can be challenging specified that the d-loop alters quickly and is subject to saturation due to unnecessary homoplasy [3]. Heterogeneity is an additional vital concern in manipulating variance time evaluations because mutations are not unsystematically dispersed through the dimension of the locus [4]. Furthermore, from d-loop sequences numerous likewise gene trees can frequently be inferred by analysing large numbers of samples [1]. By calculating single nucleotide polymorphisms (SNPs) broad studies of the human mtDNA genome have been accomplished, resolved by arrangements of the first hypervariable section of the d-loop and by examination of restriction fragment length polymorphism (RFLP) [5]. Human mtDNA is geographically arranged and can be classified into groups of associated haplotypes (haplogroups) by these studies [6]. In spite of the past arguments, the field of ancient DNA is now an effective research area due to improvements in the methodologies [2]. A series of updated studies have explained the true potential of ancient DNA samples to study the evolutionary process and to examine the models and hypothesis commonly used to rebuild the patterns of evolution and to evaluate population genetics. Recent approaches in the technologies for the analysis of DNA, such as next generation sequencing make it possible to retrieve information related to DNA archaeological remains to go back in time and study the relationships between extinct organisms and their existent relatives on genetic bases. With the advanced technologies like the next generation sequencing methodologies, researchers can decode the information even from the human degraded samples, for which the technical pitfall of classical methodologies required restrictive criteria to guarantee the authenticity of the results [7].
Molecular anthropology researchers around the world have sequenced the genomes of 1000s year’s old human bones and teeth collected from diverse areas [8]. Here we use 46 whole ancient mitochondrial genome retrieved from different servers and did some bioinformatics analysis using updated and advance tools. The ultimate goal was to use sequence data to explore and understand the position of diverse genomes using phylogenetic study.
Methods![]()
a. Sequence Selection
A detailed survey and literature review were performed to search out the ancient genomes reported by scientists around the world. More than 100 research articles were found on the PubMed who mentioned that they have shared their data for public use. Only 46 complete genomes were found and retrieved from the NCBI, GenBank and EBI in FASTA format [8]. The ages mentioned of each sample based on the publicly available papers.
b. Sequence Alignment
The sequence set was run through ClustalW, a general-purpose multi-sequence alignment of nucleic and protein sequences program [9]. ClustalW was chosen because of its robustness and reliability of the results [5]. The multiple sequence alignment was performed on high performance computer. The alignment file was generated successfully and saved for further analysis [10].
c. Phylogenetic Tree
The alignment file generated in the previous step was loaded to the BioEdit and phylogenetic analysis was done using PHYLIP. The Newick file generated was saved and MEGA 6 was then used to visualize it in different layouts. The trees produced were saved as image file.
d. Mitochondrial Haplotype Prediction
Haplotypes of each ancient mitochondrial genome was predicted using server MITOMASTER [11]. The results were obtained in tabulated format having information like haplotypes, frequency of the variants and list of variants responsible for each haplotype.
Results![]()
a. The mtDNA Sequence Set and Haplogroup-Related Polymorphisms
Around 46 complete mitochondrial ancient genomes were gathered and analysed. In these analyses, the main focus was on ancestry of these whole mitochondrial genomes where we find the roots of these ancient individuals by constructing their phylogenetic tree. The already available online ancient genomes were selected randomly and assembled using phylogenetic software where an amazing ancestry matching of some of the genomes were found where they not even close to the area they were extracted from.
b. Phylogenetic tree analysis
We aligned 46 ancient genomes, collected online and estimated trees by using neighbour-joining, maximum parsimony and maximum-likelihood. Support for nodes was assessed with bootstrap replicates. During our analysis a strong bond between genomes of Altai Neanderthal, Motala 12, Motala 1, Loschbour, Ust'-Ishim, LBK, Mezmaiskaya Neanderthal, Denisova, RISE391(ERR844272), Clovis Anzick-1, RISE395(ERR844275) and RISE210(ERR844262) was found. In this context, these ancient samples recommended the presence of a mutual earliest genomic signature. Numerous gene flow trials happened between Neanderthals, Denisovans and early modern Homo sapiens, are said to be the closest to present day humans in respect to other organisms that are revealed by study of the connections and population antiquity of existing genomes and modern human genomes. Some of these genomes were sampled from same geographic area like Altai Neanderthal, Motala 12 and Motala 1 were taken from South central-Siberia which somehow make sense to have same ancestral relation but some of them were sampled from even distinct areas like Clovis Anzick-1 from central Siberia and LBK from Sweden [12] which was quite far from their geographic range but still they showed a very strong lineage. Mitochondrial DNA genetic diversity was about one-third of that in present-day modern humans in investigation of Neandertals that existed 38,000 to 70,000 years before. These statistics collected with evaluation of mtDNA protein evolution, recommend that the enduring actual population size of Neandertals was lesser than that of present humans and existent great apes [13]. Deep ancestral relationship was found in Mal’ta, La Brana-Arintero, KO1, NE1, NE6, BR2 and NA43 (SRR1314599). Genomic analysis from the KO1, NE family and BR2, NA43 suggest no Neolithic presence in Hungarian Neolithic period. Here too the samples taken were not even close in their geographic range but they shared a strong ancestral relationship like KO1 were taken from Hungry [14] which were said to be of Neolithic period but it’s very close ancestral lineage companion La Brana-Arintero were sampled from Spain but these samples shared same ancestors with each other. The ancestral Impact of RISE98 (ERR844252), RISE446 (ERR844288), RISE569 (ERR844336) and RISE97 (ERR844251) were checked and revealed that they share same ancestral lineage. Analysis of the relationships of RISE174 (ERR844258), RISE496 (ERR844301), RISE21 (ERR844245), RISE509 (ERR844311), RISE94 (ERR844250), RISE150 (ERR844256), RISE240 (ERR488263), RISE479 (ERR844290) and Bot15 (ERR668415) also shared same ancestral lineage. The RISE family were sampled across Eurasia but there was genome of Bot15 (ERR668415) which were humans from Bronze Age when compared to present day human [15]. Beside similarities of these genome some of them showed a weaker support in sharing same ancestors. The Altai Neanderthal and Bot15 (ERR668415) both were sampled across Eurasia but do not share any ancestral relationship (Figure 1).
Tables & Figures
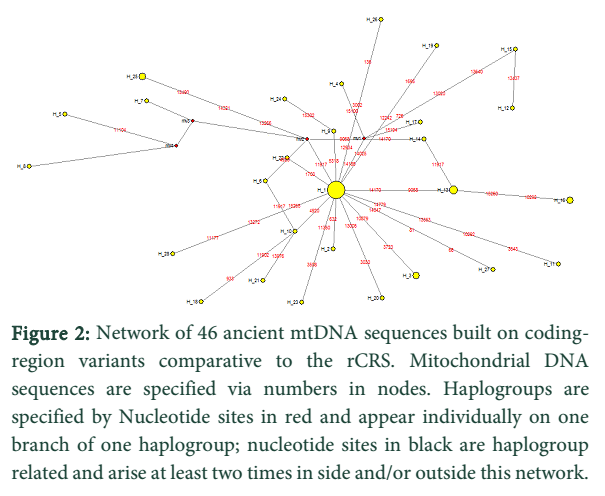
Discussion![]()
The relation obtained contributes to a very huge genetic shift and lake of gene flow [15]. Additionally, an absolute list of substitutions that developed in present humans after split-up from the descendants of Neanderthals and Denisovans, was established by the high-grade Neanderthal genome. We also found that KOS 14 comprises additional Neanderthal DNA that is restricted in extended territories than present-day human where KOS is European Russia dating from 38,700 to 36,200 years ago [16]. An era of most important ethnic fluctuations was the RISE family form Eurasia about 3000–1000 BC, the exchange of concepts or since human migrations, possibly more over enabling the spread of languages and definite phenotypic characters were accountable for these variations. A significant population immigrations and alternates, accountable for influencing main parts of current demographic structure together in Europe and Asia is supported by the Bot15 (ERR668415) and RISE family. In the initial bronze period, ancestral similarity among these populations also share the theorised blow-out of Indo-European languages [17]. Mechanisms of pathogen development and alteration for evolving and reappearing toxicities is also explained by this study. We aim that this study will help researchers in understanding the evolutionary position of ancient populations resided around the world.
Acknowledgment
We are thankful to Dr. Muhammad Ilyas, Assistant Professor at Centre for Human Genetics Mansehra for his generous help and support during this project.
The authors declare that there is no conflict of interest regarding the publication of this paper.
References![]()
- Ballard JWO, Whitlock MC. The incomplete natural history of mitochondria. Molecular ecology, (2004); 13(4): 729-744.
- Salas A, Richards M, Lareu M-V, Scozzari R, Coppa A, et al. The African diaspora: mitochondrial DNA and the Atlantic slave trade. The American Journal of Human Genetics, (2004); 74(3): 454-465.
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular biology and evolution, (1993); 10(3): 512-526.
- Excoffier L, Yang Z. Substitution rate variation among sites in mitochondrial hypervariable region I of humans and chimpanzees. Molecular biology and evolution, (1999); 16(10): 1357-1368.
- Chen X, Oh S-W, Zheng Z, Chen H-W, Shin H-h, et al. Cyclin D-Cdk4 and cyclin E-Cdk2 regulate the Jak/STAT signal transduction pathway in Drosophila. Developmental cell, (2003); 4(2): 179-190.
- Chen J, Farrell JW, Murray DW, Prell WL. Timescale and paleoceanographic implications of a 3.6 my oxygen isotope record from the northeast Indian Ocean (Ocean Drilling Program site 758). Paleoceanography, (1995); 10(1): 21-47.
- Whittaker ET, Watson GN A course of modern analysis. 1996; Cambridge university press.
- Maddison W. Analysis of phylogeny and character evolution. MacClade, (1992); 53(1-4):190-202.
- Vigilant L, Stoneking M, Harpending H, Hawkes K, Wilson AC. African populations and the evolution of human mitochondrial DNA. Science, (1991); 253(5027): 1503-1507.
- McWilliam H, Valentin F, Wallace I, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics 23: 29472948Leake JR (1994) Tansley Review No. 69. The biology of mycoheterotrophic (saprotrophic) plants. New Phytologist, (2007); 127171216Lee.
- Cann RL, Stoneking M, Wilson AC. Mitochondrial DNA and human evolution. Nature, (1987); 325(6099): 31.
- Lazaridis I, Patterson N, Mittnik A, Renaud G, Mallick S, et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature, (2014); 513(7518): 409.
- Briggs AW, Good JM, Green RE, Krause J, Maricic T, et al. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science, (2009); 325(5938): 318-321.
- Gamba C, Jones ER, Teasdale MD, McLaughlin RL, Gonzalez-Fortes G, et al. Genome flux and stasis in a five millennium transect of European prehistory. Nature communications, (2014); 55257.
- Allentoft ME, Sikora M, Sjögren K-G, Rasmussen S, Rasmussen M, et al. Population genomics of bronze age Eurasia. Nature, (2015); 522(7555): 167.
- Seguin-Orlando A, Korneliussen TS, Sikora M, Malaspinas A-S, Manica A, et al. Genomic structure in Europeans dating back at least 36,200 years. Science, (2014); 346(6213): 1113-1118.
- Achilli A, Rengo C, Magri C, Battaglia V, Olivieri A, et al. The molecular dissection of mtDNA haplogroup H confirms that the Franco-Cantabrian glacial refuge was a major source for the European gene pool. The American Journal of Human Genetics, (2004); 75(5): 910-918.
![]()
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0