

Full Length Research Article
Genotypic and computational sequence analysis of ALADIN gene causing Allgrove syndrome
Fezza Arshad1, Nida Abdul Qadir1*, Nosheen Ishaq1, Saqib Mehmood2, Aiman Shehzad1, Warda Fatima1
Adv. life sci., vol. 7, no. 1, pp. 10-15, November 2019
*– Corresponding Author: Nida Abdul Qadir (Email: nida.aq234@gmail.com)
Authors' Affiliations
2. University of Health Sciences, Lahore – Pakistan
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Allgrove syndrome is autosomal recessive disorder, the gene involved in this syndrome is known as ALADIN located close to type 2 keratin gene cluster on chromosome 12q13 whose function is to control the nucleocytoplasmic trafficking and also affects the nuclear pore complexes. Mutation in this gene cause triple A syndrome. The aim of research was to analyze the mutational changes in ALADIN gene, formation of 3D Structure of normal and mutated protein and differentiation of normal and mutated protein.
Methods: Genotyping by using tetra arm PCR and Sequence analyses of coding region of ALADIN gene was done in two families having affected children with Allgrove’s syndrome.
Results: Point mutation in exon 1 and alteration in 3D structure of protein was observed by using VMD (Visual molecular dynamics) that shows truncation, absence of few amino acid and structural modification of proteins which alters in transportation ability.
Conclusion: It is concluded from the study that proper structure and function of NPC (nuclear pore complex) binding protein is necessary in normal body function and if any mutation is present in ALADIN gene it can cause symptoms of rare Allgrove’s syndrome.
Keywords: ALADIN gene; 3D protein structure; Triple A syndrome
Introduction![]()
Allgrove syndrome or triple A syndrome (AAAS) is a hereditary trait acquired with the autosomal recessive inheritance which is depicted in a triad of adrenal deficiency, alacrima and achalasia. One A refers to Achalasia, known as esophageal aperistalsis and damage to lower esophageal sphincter (LES) because of loss of inhibitory neurons in the esophageal myenteric plexus that results in failure of sphincter muscles to relax [1]. Achalasia is a disorder that influences the ability to move nourishment through the throat, the tube that conveys food from the throat to the stomach. It can cause extreme nourishing problems and low glucose level in body a condition known as hypoglycemia [2]. Second A refers to Adrenal deficiency also known as Addison disease. Adrenal deficiency occurs when adrenal glands do not produce sufficient amount of hormone known as cortisol. Cortisol activates the body in stress condition. Third A refers to alacrima, which is congenital in origin includes a vast range of lacrimal secretory disorders. Patients with alacrima show complete absence of tears to lower secretion of tears (hypolacrima). Alacrima-achalasia disorder with the absence of adrenal deficiency is depicted [3]. Adrenal deficiency in people with Allgrove syndrome starts in younger age but it can also start later in life during their 30s [4]. Patients with Allgrove syndrome generally show hypoglycemia because of adrenal deficiency. Individuals with triple A syndrome regularly encounter strange sweating, trouble directing pulse, anisocoria, dysautonomia, developmental delay, ataxia and polyneuropathy with sensory, motor and autonomic components, mild dementia and long-tract degeneration and parkinsonism [5, 6]. Roubergue et al., reported three children with myoclonus of upper limbs and face, extensive GIT dysfunction as well as dysmotility showing small gall bladder producing small bowels [7]. According to Thummler et al., in some patient’s medical history is marked by achalasia and mega esophagus. Confirmation of Clinical diagnosis is done by molecular analysis of the AAAS gene on chromosome 12q13 [8]. Allgrove syndrome is diagnosed in 100 people worldwide showing autosomal recessive pattern of inheritance [9]. Allgrove syndrome was considered as an alternative of (FGD) familial glucocorticoid deficiency because of appearance of ACTH insensitivity in the couple of disorders, while no mutation was observed in ACTH receptor gene (18p11.2) causing FGD [10]. Weber et al., in 1996 successfully located the gene for Allgrove syndrome close to type 2 keratin gene cluster on chromosome 12q13 [6]. Handschug et al., in 2001 discovered 16 exons of the gene and named it as ALADIN. ALADIN is actually a protein performs functions like protein to protein interaction, vesicular transportation, RNA development, signal transduction, cell division management and cytoskeleton assembly includes in a section of mammalian NPC (nuclear pore complex). NPC is essential in connecting cytoplasm and nucleus of different cells. As a result of mutation, ALADIN gene dissocialized to cytoplasm instead of NPC, while microscopic analysis of Allgrove patients showed that triple A syndrome is not a structural disorder at cellular level [11, 12]. The genetic map of ALADIN gene is on chromosome 12q13, it contains 16 exons and encodes 547 amino acids located near type II keratin gene clusters and some other potential gene are also present in nearby region named as SCN8A and HOXC genes [13]. In this syndrome, various homozygous and compound heterozygous mutations are reported. Most prevalent mutation a splice donor mutation IVS14+1G→A is observed in all patients. Another prominent novel splice donor mutation IVS11+1G→A is observed in exon 11 of AAAS patients [14]. Collectively nine different types of ALADIN mutations are reported 2 nonsenses, 5-point mutations and 2 frameshifts. Four-point mutations are observed one is near to the N terminus of ALADIN gene while rest of the three is present in the WD-repeat domain. Various disease-correlated missense, frameshift and nonsense mutations failed to show any effect on the structure of Nuclear Pore Complexes however functional irregularities are observed. No apparent or visual irregularities of the cores, NPCs and nuclear envelopes are observed by analysis of cells taken from Allgrove patient [15]. In a Pakistani pedigree homozygous nonsense mutation in GMPPA (guanosine diphosphate (GDP)-mannose pyro phosphorylase A) showed autonomic dysfunction, intellectual disability, alacrima, achalasia, gait flaws, and delayed development. This autosomal recessive disorder depicted resemblance to Allgrove syndrome. It is reported that a novel R155P mutation in ALADIN gene is linked with Allgrove syndrome and insulin-activated hypoglycemia, detected by ACTH stimulation tests [16].
Methods![]()
Collection of blood sample:
Patients with Allgrove syndrome were identified from hospital having clinical data record and 5cc venous blood was collected from patient after informed consent from parents of patients. Sample were collected in 0.5 M EDTA and stored at -4oC until further processed.
DNA Isolation and Quantification:
All the samples of Allgrove syndrome patients and healthy controls were processed and DNA is isolated by using manual techniques and by kit method following the manufacturer’s instructions. Isolated DNA samples were stored at -20oC or 4oC as required. DNA samples were resolved by running the 2% TBE agarose gel along with the standard DNA (DNA of known concentration) after mixing with 6X loading dye. Gel was observed under ultraviolet light (by using WEALTEC UV-transilluminator). Quantification was done by comparing the intensity of DNA samples with the standard.
Primer designing, PCR and Sequencing:
Primers of ALADIN gene were designed by software primer3. Primers were optimized according to their respective melting temperature Tm. For exon 1 (58°C), exon 2 (58°C), exon 3 (57°C) exon 4 and 5 (58°C), exon 6 (56°C), exon 7 (57°C), exon 8 (58°C), exon 9 (56°C), exon 10 and 11 (57°C), exon 12 and 13 (59°C) exon 14 and 15 (58.5°C ), exon 16 (57°C). These 16 exon of the ALADIN gene were amplified by PCR. The intronic regions between the exons 4and 5, 10 and 11, 12 and 13, and 14 and 15 were small enough to allow these exons to be amplified together. PCR was carried out and PCR fragments were observed on a 1% agarose gel. Big dye terminator cycle sequencing ready reaction Kit was used to sequence the PCR products of ALADIN gene. Chromatograms from normal and effected individuals were compared with the corresponding control gene sequence from NCBI to identify the aberrant nucleotide base pair change.
Results![]()
Sequence analysis:
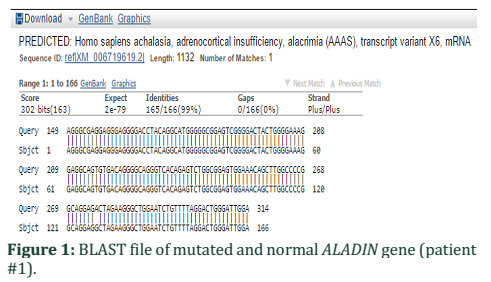
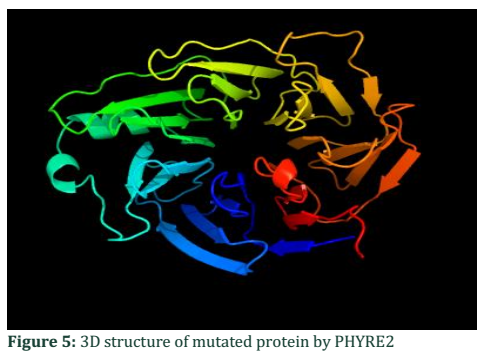
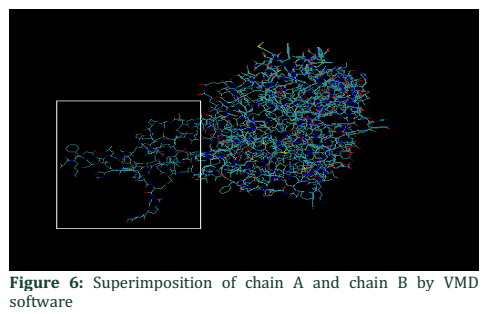
The present study involved the sequence analysis of coding regions of ALADIN gene in two families having affected children with Allgrove syndrome. When the sequences of two patient samples were BLAST with normal gene sequence present in NCBI a mismatch in exon 1 of patient #1 was found (Figure 1). Analysis of chromatogram sequence reveal the presence of nucleotide A instead of nucleotide G at position number 128 (figure 2 and 3). This mutation 128G>A was found to substitute the threonine with alanine and causes immature truncation of mutated protein. Figure 4 shows the normal while (Figure 5) shows the mutated protein structure. Immature truncation of protein is shown in (Figure 6) whole protein is unable to superimposed with normal protein, and the sequence of 5aa is missing. In the other patient studied no mutation in any exon was found.
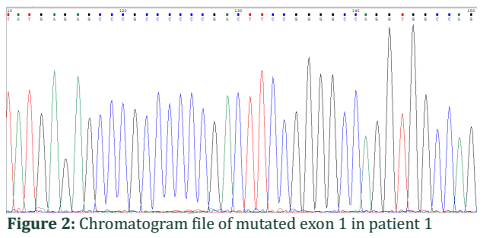
Chromatogram file of mutated exon 1 in patient 1:
The mutated gene has G nucleotide at 128 position of exon 1 showing a black peak in sequenced results (Figure 2).
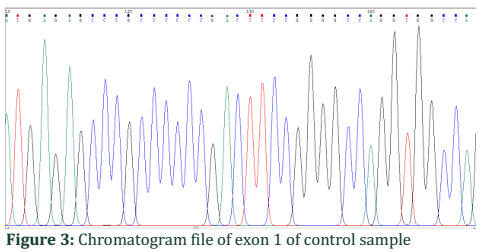
Chromatogram file of exon 1 of control sample:
The normal gene has nucleotide A at 128 positions of exon 1 shown by a green peak in sequenced results (Figure 3).
3D Structures of protein:
The 3D structure is obtained by incorporating FASTA sequence of ALADIN protein retrieved from NCBI into a software PHYRE2. Scenario of this software is web entryway for protein displaying, determination and examination. By comparing both chromatogram files of patient and control it is able to distinguish the peak pattern, color change and dimensions.
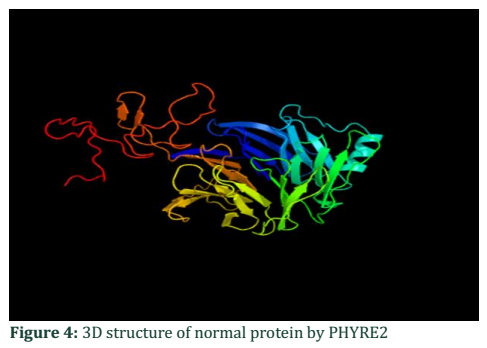
3D Structure of ALADIN gene of normal protein (Chain A):
FASTA format of amino acid sequence retrieved from NCBI. Here threonine highlighted in greenish normally present. MCSLGLFPPPPPRGQVTLYEHNNELVTGSSYESPPPDFRGQWINLPVLQLTKDPLKTPGRLDHGTRTAFIHHREQVWKRCINIWR
VGLFGVLNEIANSEEEVFEWVKTASGWALALCRWASSLHGSLFPHLSLRSEDLIAEFAQVTNCTIVPSLKHRLQRNVASLAWKPLSA
SVLAVACQSCILIWTLDPTSLSTRPSSGCAQVLSHPGHTPVTSLAWAPSGGRLLSASPVDAAIRVWDVSTETCVPLPWFRGGGVTN
LLWSPDGSKILATTPSAVFRVWEAQMWTCERWPTLSGRCQTGCWSPDGSRLLFTVLGEPLIYSLSFPERCGEGKGCVGGAKSATI
VADLSETTIQTPDGEERLGGEAHSMVWDPSGERLAVLMKGKPRVQDGKPVILLFRTRNSPVFELLPCGIIQGEPGAQPQLITFHPSF
NKGALLSVGWSTGRIAHIPLYFVNAQFPRFSPVLGRAQEPPAGGGGSIHDLPLFTETSPTSAPWDPLPGPPPVLPHSPHSHL
3D Structure of ALADIN gene of mutated protein (Chain B)
FASTA format of amino acid sequence is retrieved from NCBI. Here A (alanine) shown in red is mutated is present in place of threonine.
MCSLGLFPPPPPRGQVTLYEHNNELVAGSSYESPPPDFRGQWINLPVLQLTKDPLKTPGRLDHGTRTAFIHHREQVWKRCINIWRD
VGLFGVLNEIANSEEEVFEWVKTASGWALALCRWASSLHGSLFPHLSLRSEDLIAEFAQVTNCTIVPSLKHRLQRNVASLAWKPLSA
SVLAVACQSCILIWTLDPTSLSTRPSSGCAQVLSHPGHTPVTSLAWAPSGGRLLSASPVDAAIRVWDVSTETCVPLPWFRGGGVTN
LLWSPDGSKILATTPSAVFRVWEAQMWTCERWPTLSGRCQTGCWSPDGSRLLFTVLGEPLIYSLSFPERCGEGKGCVGGAKSATI
VADLSETTIQTPDGEERLGGEAHSMVWDPSGERLAVLMKGKPRVQDGKPVILLFRTRNSPVFELLPCGIIQGEPGAQPQLITFHPSF
NKGALLSVGWSTGRIAHIPLYFVNAQFPRFSPVLGRAQEPPAGGGGSIHDLPLFTETSPTSAPWDPLPGPPPVLPHSPHSHL
This change in amino acid is manually done using FASTA sequence of protein as we know the change by sequencing.
Superimposition of chain A and chain B
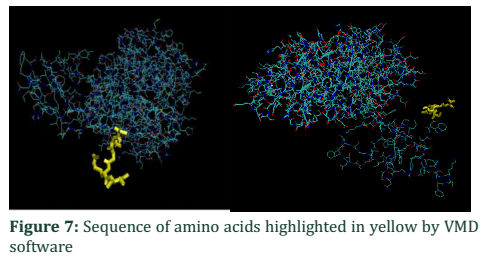
In order to analyze the difference between two chains VMD and FATCAT structure alignment software is applicable to visualize the structural change between them. Most of the structure in Figure 6 showed structure alignment except that shown in the box which is a part of normal chain A. For detailed analysis of amino acid sequence VMD is used, the sequence viewer showed that chain A has 460aa while that of B has 455aa. Figure 7 showed the amino acid sequence highlighted in yellow unable to super imposed with each other, part of chain A. Due to this difference of 5aa, some of the part remain free from compact globular structure.
Figures & Tables
Discussion![]()
Allgrove syndrome is a hereditary trait acquired as an autosomal recessive condition which is portrayed in a trine of adrenal inadequacy, alacrima and achalasia. AAAS is an abbreviated form of Allgrove syndrome. Adrenal insufficiency involves inappropriate functioning of adrenal glands present in proximal part of kidney, alacrima refers to the absence of tears during cry of a child because tears secreting glands lose their functioning as the transportation of cell is affected that propagate tears out of the cell and the last one is achalasia that involves the improper functioning of sphincter muscles present on the distal end of esophagus and stomach, when food is passed through the muscle it is unable to hold that bolus [16, 17].
The main focus of the study was to determine mutations in coding region of ALADIN that might cause this syndrome, the product of ALADIN gene acts as subunit of nuclear pore complex involved in transportation, role in the formation of mitotic spindle during cell division, controls hypoglycemia, in fact it is involved in multiple sets of processes. Different types of mutations are reported from different regions of world. But in this study, we found a point mutation in exon1 that alters the amino acid sequence in protein polypeptide which was not similar in both patients. The mutation that is observed in present study was similar to the mutation found in an article published in 2009 that transforms A to G in exon 1 showed ophthalmic abnormalities [18]. It was reported that clinical diagnosis of Allgrove patients is difficult as it is an autosomal recessive trait, secondly unable to know the complete history of patients; thirdly different patients show only two or more symptoms of this disease.
Alteration which is observed in protein was the modification of its structure that alters the carrying or transporting ability of mutated protein. The protein structure of both of the patient 1 and control were different and are not completely superimposed on each other concluding this patient shows different morphology of protein structure. The mutated protein having point mutation in exon 1 showed the absence of 5 amino acids in its polypeptide chain and the second patient was used as control in order to determine or estimate the appeared differences. The results of patient 2 were different as no mutation in all the exons was observed. Similarly, no mutation in ALADIN gene is also reported in Allgrove patients showing genotypic heterogeneity [19]. It is concluded from the study that ALADIN gene should performs its function properly in NPC binding which is necessary for normal body functioning and if any alteration/ mutation is present in ALADIN gene it can cause symptoms of rare disorder AAA syndrome. Although further studies are required to find actual cause of AAAS.
In this study found a point mutation in exon1 that alters the amino acid sequence in protein which was not similar in both patients of triple A syndrome. Findings of the present study seem to be interesting and analysis on larger cohort needs to be performed to validate the results.
Authors' Contribution
Fezza Arshad performed the experiment and prepared figures. Nida Abdul Qadir authored and reviewed draft of paper. Nosheen Ishaq performed the experiment. Muhammad Aiman Shehzad identified the patients. Saqib Mehmood identified the patients. Warda Fatima supervised, reviewed and approved the final draft of paper.
Authors' Contribution
The authors would like to thank all volunteers for their participation in this study. The authors also express their gratitude to the Department of Chemistry, University of Azad Jammu and Kashmir, Muzaffarabad, Pakistan, for providing required facilities.
The authors declare that there is no conflict of interest regarding the publication of this paper.
References![]()
- Gockel HR, Schumacher J, Gockel I, Lang H, Haaf T, et al. Achalasia: will genetic studies provide insights? Human Genetics, (2010); 128(4): 353-64.
- Kunte H, Nümann A, Ventz M, Siebert E, Harms L. Wernicke’s encephalopathy in a patient with triple A (Allgrove) syndrome. Journal of neurology, (2011); 258(10): 1882-4.
- Menon SK, Bangar TR, Kaba A, Shah R, Menon PS, et al. Triple A syndrome. Indian Journal Pediatrics, (2008);75(9):967-.
- Houlden H, Smith S, de Carvalho M, Blake J, et al. Clinical and genetic characterization of families with triple A (Allgrove) syndrome. Brain, (2002); 125(12): 2681-90.
- Nakamura K, Yoshida K, Yoshinaga T, Kodaira M, Shimojima Y, et al. Adult or late-onset triple A syndrome: case report and literature review. Journal of the Neurological Sciences, (2010); 297(1): 85-8.
- Dumić M, Barišić N, Rojnić-Putarek N, Kušec V, Stanimirović A, et al. Two siblings with triple A syndrome and novel mutation presenting as hereditary polyneuropathy. European Journal of Pediatrics, (2011); 170(3): 393-6.
- Roubergue A, Apartis E, Vidailhet M, Mignot C, Tullio‐Pelet A, et al. Myoclonus and generalized digestive dysmotility in triple A syndrome with AAAS gene mutation. Movement disorders: official journal of the Movement Disorder Society, (2004); 19(3): 344-6.
- Thümmler S, Huebner A, Baechler-Sadoul E. Triple A syndrome: two novel mutations in the AAAS gene. BMJ case reports, (2009): bcr0920080984.
- Bizzarri C, Benevento D, Terzi C, Huebner A, Cappa M. Triple A (Allgrove) syndrome: an unusual association with syringomyelia. Italian Journal of Pediatrics, (2013); 39(1): 39.
- Clark AJ, Cammas FM, Watt A, Kapas S, Weber A. Familial glucocorticoid deficiency: one syndrome, but more than one gene. Journal of Molecular Medicine, (1997); 75(6): 394-9.
- Weber A, Wienker TF, Jung M, Easton D, Dean HJ, et al. Linkage of the gene for the triple A syndrome to chromosome 12q13 near the type II keratin gene cluster. Human Molecular Genetics, (1996); 5(12): 2061-6.
- Dixit A, Chow G, Sarkar A. Neurologic presentation of triple A syndrome. Turkiye Klinikleri Journal Pediatrics, (2011); 45(5): 347-9.
- Handschug K, Sperling S, Yoon SJ, Hennig S, et al. Triple A syndrome is caused by mutations in AAAS, a new WD-repeat protein gene. Human Molecular Genetics, (2001); 10(3): 283-90.
- Koehler K, Malik M, Mahmood S, Gießelmann S, Beetz C, et al. Mutations in GMPPA cause a glycosylation disorder characterized by intellectual disability and autonomic dysfunction. The American Journal of Human Genetics, (2013); 93(4): 727-34.
- Kind B, Koehler K, Krumbholz M, Landgraf D, Huebner A. Intracellular ROS level is increased in fibroblasts of triple A syndrome patients. Journal of Molecular Medicine, (2010); 88(12): 1233-42.
- Hirano M, Furiya Y, Asai H, Yasui A, Ueno S. ALADINI482S causes selective failure of nuclear protein import and hypersensitivity to oxidative stress in triple A syndrome. Proceedings of the National Academy of Sciences, (2006); 103(7): 2298-303.
- Spiegel R, Shalev S, Huebner A, Horovitz Y. Association of chronic symptomatic neutropenia with the triple A syndrome. Journal of Pediatrics, hematology/oncology, (2005); 27(1): 53-5.
- Villanueva-Mendoza C, Martinez-Guzman O, Rivera-Parra D, Zenteno JC. Triple A or Allgrove syndrome. A case report with ophthalmic abnormalities and a novel mutation in the AAAS gene. Ophthalmic Genetics, (2009); 30(1): 45-9.
- Palka C, Giuliani R, Brancati F, Mohn A, Di Muzio A, Calabrese O, Huebner A, De Grandis D, Chiarelli F, Ferlini A, Stuppia L. Two Italian patients with novel AAAS gene mutation expand allelic and phenotypic spectrum of triple A (Allgrove) syndrome. Clinical Genetics, (2010);77(3):298-301.
![]()
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0