

Full Length Research Article
Mutational analysis of CYP1B1 gene in Pakistani pediatric patients affected with Primary Congenital Glaucoma
Muhammad Umer Khan1,2,*, Raima Rehman3, Haiba Kaul3, Saqib Mahmood3, Ali Ammar3
Adv. life sci., vol. 7, no. 1, pp. 32-37, November 2019
*– Corresponding Author: Muhammad Umer Khan (Email: umer.khan685@gmail.com)
Authors' Affiliations
2. The University of Lahore, Pakistan
3. University of Health Sciences, Lahore, Pakistan
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Glaucoma is the significant cause of blindness all over the world. Primary congenital glaucoma (PCG) reduces the vision and ultimately causes the blindness by damaging the aqueous drainage system of the eye. The purpose of the current study was to determine the pathogenic mutations in the CYP1B1 gene responsible for PCG.
Methods: A total of thirty-five PCG patients were enrolled in this study. Blood samples were collected from the enrolled patients, and after DNA extraction and amplification, the coding regions of CYP1B1 were sequenced to determine the pathogenic mutations. In-silico analysis of the identified mutation was executed to study the effect of genetic variation on protein structure.
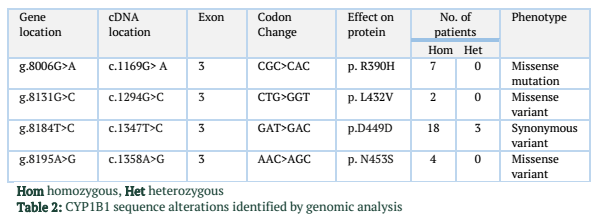
Results: One mutation, c.1169 G>A has been revealed in exon 3 of the CYP1B1 gene leading to p.R390H, present in 20% of the patients enrolled. Besides, two missense sequence variants c.1294G>C (2 patients), c.1358A>G (4 patients) and a synonymous variant c.1347T>C (18 patients) has also been observed.
Conclusion: Our study not only reaffirms the role of CYP1B1 mutations in PCG but also supports the use of genetic screening for molecular diagnosis and carrier identification, which will reduce the burden of disease on society. Furthermore, the in-silico analysis of the identified mutations provided an in-depth understanding of the PCG pathogenesis at the molecular level.
Keywords: Primary congenital glaucoma; CYP1B1, mutation; Genetic variation
Introduction![]()
It is assessed that glaucoma causes the blindness or vision impairment in 6.9 million individuals while the overall 2.2 billion people have blindness [1]. It is a disorder in which death of retinal ganglion cells (RGC’s) occurs, usually due to high intraocular pressure, which leads to the atrophy of the optic nerve and results in gradual visual field damage and ultimately irreversible vision loss [2]. In general, primary congenital glaucoma (PCG), primary open-angle glaucoma (POAG) and primary angle-closure glaucoma (PACG) are the three main types of glaucoma [3]. Primary congenital, or infantile, glaucoma has onset within the one year of life. Approximately, it happens 1 out of 10,000 births, and reduced vision occurs in 10 percent cases [4]. According to a prevalence prediction study, it is estimated that over 11.1 million people will be bilaterally blind with PCG by 2020 [5].
PCG is a condition without a cure that manifests itself in the recessive form. According to the data figures, it is estimated that nearly 13.5% of cases of blindness are due to glaucoma worldwide [6]. Primary open-angle glaucoma (POAG) occurs most commonly among different subtypes of glaucoma and causes more than 50% of the total glaucoma cases worldwide [7]. Primary congenital glaucoma (PCG) accounts for 0.01-0.04% cases of blindness all over the world [8]. PCG is usually rare but is the common form of glaucoma in children often manifesting within the first three years of life. It has been observed that PCG is more prevalent in males as compared to females (65% versus 35%, respectively) and in 60-80% of cases patient’s both eyes are affected [3]. PCG has higher incidence rate in families where consanguinity is accustomed and in genetically inbred populations [9,10]. PCG is inherited in autosomal recessive mode, but some cases of pseudo dominance are also reported [11].
The pathophysiological mechanism of PCG is not fully understood. Increased IOP could compress the optic nerve at the lamina cribosa, block the axoplasmic flow, and interfere in retrograde transport of neurotrophin to RGCs [12]. Elevated IOP could also affect capillary blood supply to the region, and all these events could progressively lead to the apoptotic death of RGC’s. With Mendelian linkage approaches and mostly affected pedigrees, autosomal recessive PCG has been mapped to four chromosomal loci: GLC3A (2p21), GLC3B (1p36) GLC3C (14q24.3) and 14q24.2-q24.3 [12-15]. Studies have reported CYP1B1 as a causal gene in PCG, and its mutations are reported in 27% of sporadic and 87% of familial cases of PCG [16]. The CYP1B1 gene is present at position 22.2 on the short (p) arm of chromosome 2, comprising of 3-exons encoding 543 amino acids [3]. CYP1B1 protein belongs to CYP450 superfamily of hemoproteins and is a membrane-bound monooxygenase with multiple functions [17]. It is present predominantly in the trabecular meshwork and neuroretina but also other tissues such as adrenal gland, kidney, uterus, and genitals both in adult and fetal life [18]. The protein is probably linked with the metabolism of those compounds that are critical for the development of the eye, but its exact role is still not known [19]. In the human eye, relatively high levels of CYP1B1 mRNA have been observed in the ciliary body and iris than retina, cornea, and retinal pigment epithelium [20]. This study was designed to find out the mutations of the CYP1B1 gene in PCG patients.
Methods![]()
It is a descriptive study performed at the Biochemistry Department of University of Health Sciences, Lahore, during the period of March to December 2014. Thirty-five PCG affected patients were recruited to participate in this study after the approval from the ethical committee of the institutional review board. Consent forms have been filled by the guardian of the patients. Complete medical history of all the patients was obtained. Ophthalmologists of Layton Rahmatullah Benevolent Trust (LRBT) and Children Hospital confirmed the diagnosis of PCG. Blood samples of all the PCG affected individuals were collected in EDTA vials. DNA was extracted by a modified nonorganic method [21,22]. Four sets of primer pairs were designed to cover the two coding exons of CYP1B1 gene by using Primer 3 genome Browser (Table 1).
Amplification was performed in a 25μl reaction containing 50ng of genomic DNA, 2.5μl 1X PCR buffer, 8pmoles of each primer, 2.5 mMdNTP, 2.5mM MgCl2, and 0.2U Taq DNA polymerase (Applied Biosystems). GeneAmp PCR System 9700 (Applied Biosystems) was used to amplify the desired region of CYP1B1 gene. Initial denaturation was done at 96°C for 7 minutes while 30 cycles (denaturation 95°C for 30s, annealing 58°C for 45s, extension 72°C for 30s) were performed and a final extension of 10 minutes at 72°C was also done.
Agarose gel (2%) was used to visualize the PCR products along with 100 bp molecular weight marker (ladder). Ethanol precipitation was done to purify the PCR product. The amplified DNA fragments were sequenced by fluorescence-based chain terminator (dideoxy) sequencing method on an ABI 3100 Automated sequencer (Applied Biosystems). Finch Tv software was used to analyze the sequencing data. To look out for sequence alterations, the sequence of each exon from all the patients was blast against the normal CYP1B1 gene sequence using BLAST 2 (Basic Local Alignment Search Tool) sequences server on NCBI website (http://www.ncbi.nlm.nih.gov/blast/bl2seq/ wblast2.cgi).
Statistical analysis
Demographic and clinical data of all PCG patients were entered using PASW-22 (SPSS Inc. Chicago USA). Continuous variables were expressed as Mean ± SD; whereas categorical variables were in the form of frequency and percentages. The amplified DNA was sequenced, and the data was analyzed by using Chromas Lite v2.01 software. The sequence of each exon was blast against the normal reference sequence of SCN5A by using BLAST (Basic Local Alignment Search Tool) sequence server on NCBI website.
Results![]()
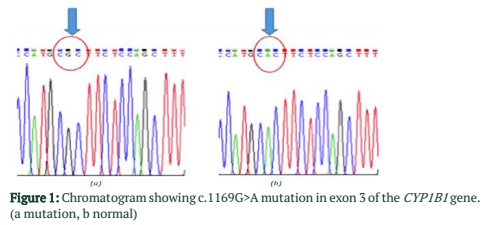
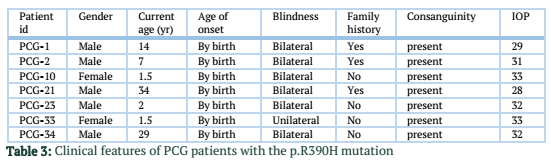
Out of 35 patients enrolled in this study, 21 (60%) were male, and 14 (40%) were female. Mean age of patients was 9.61 ± 13.9 years (Mean ± S.D). Among them, 31 (89%) patients were bilaterally affected by PCG, whereas the rest of the 4 (11%) patients were affected unilaterally. No eye abnormality other than PCG was associated with these patients. Family history of PCG was present in 14 patients (60%). Consanguinity was found in 30 patients (86%). Results of DNA sequencing for CYP1B1 gene revealed four sequence variations in the coding region of CYP1B1 gene. All of these variations were found in exon 3. All the variations reported in this study are shown in table 2. Seven patients (20%) revealed a mutation c.1169 G>A in exon 3 of CYP1B1 gene. Five of them were male, and two were female. All of these patients had consanguineous parents, and three of them showed a positive family history. This mutation altered the sequence of codon number 390 CGC for Arginine to CAC resulting in a Histidine creating a missense mutation (p.R390H). Figure 1 shows the representative chromatogram showing c.1169G>A mutation in exon 3 of the CYP1B1 gene.
A substitution of T>C was observed at nucleotide position c.1347 in exon 3 of the CYP1B1 gene. However, this change in DNA nucleotide sequence did not result in an amino acid change (GAT>GAC Aspartic acid>Aspartic acid). It is polymorphism and was found in homozygous state in 18 patients (51%) in this study. Also the same polymorphism in heterozygous condition was found in 3 patients (8%). Another SNP (single nucleotide polymorphism) AAC-to-AGC was found at position c.1358 in four cases. It replaced codon number 453 for Asparagine (AAC) into a Serine (AGC) resulting in a polymorphism. Sequencing analysis of exon 3 of the CYP1B1 gene showed another polymorphism in two patients (6%). This change occurred due to C>G at position c.1295 resulting in codon 432 CTG-to-GTG (Leu-to-Val).
In silico analysis
In silico analysis has been performed to determine the pathogenic effect of p.R390H mutation. SIFT software was used to evaluate the effect of c.1169 G>A mutation on protein function (http://sift.jcvi.org/). It revealed a score of 0.0001 for p.R390H. The SIFT score of less than 0.05 showed the pathogenic effect of the mutation.
Therefore, it is suggested that substitution at codon 390 from R to H is harmful for the protein function. So SIFT postulated that p.R390 has a vital role in CYP1B1 protein structure and ultimately its proper function.
Clustalw2.1 (http://www.ebi.ac.uk/Tools/services) was used to perform the multiple sequence alignment of CYP1B1 among various species. Arginine at position 390 is found to be conserved in the CYP1B1 protein among different species, emphasizing its importance in protein molecule (figure 2).
PolyPhen2 was also used to predict the pathogenic role of c.1169G>A mutation which was based on physical and comparative considerations. (http://genetics.bwh.harvard.edu/cgibin/ggi/ggi2.cgi). This web tool predicted this mutation to be pathogenic with a score of 1.00 (figure 3).
Model building and structure analysis
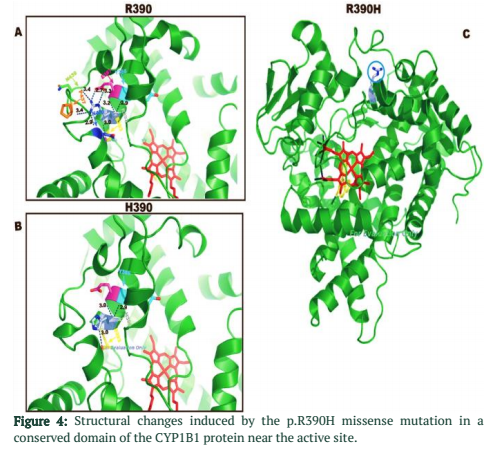
A 3D model was built to study the protein-level mechanisms of the p.R390H mutation for a bioinformatics structural analysis by using I-TASSER (Figure 4).
A ribbon diagram of the CYP1B1 protein is shown on the right with heme group of the active site in red sticks while ligand (α-Naphthoflavone) in the yellow sticks with polar contacts as blue circle indicates the location of the p.R390H mutation. Inserts A and B compare the H-bonding situation and resultant conformational changes due to the p.R390H mutation. Arg390 lies close to the active site of CYP1B1 enzyme, its substitution by the His results in the loss of structuring H-bonds (dotted lines) with Glu387, Asn428, Pro437, and Asn439. Amino acids involved in hydrogen bonding are shown as sticks (distances also indicated) with C atoms for R390 in grey, N atoms in blue, O atoms in red and S atoms in orange respectively.
R390H cause the parallel orientation of the Glu387 and His390 residues side-chain-interaction to change to the end of Asn428 in mutated form while the helical K domain of CYP1B1 protein in wild type showed the analogous arrangement of side chains of Glu387 and Arg390. It forms a hydrogen-bonding interaction with the turn-apart meander region end of Asn428, so it is considered that this structure is required for haem binding and overall proper functioning of the molecule.
Figures & Tables



Discussion![]()
Primary congenital glaucoma is due to the pathogenic mutations of CYP1B1 [3]. In the current study, out of 35 PCG patients enrolled, 21(60%) were male, and 14(40%) were female. These results are following the results of a study conducted by Vasiliou and Gonzalez, who reported that PCG is more prevalent in males (65%) as compared to females (35%) [3]. Frequency of PCG causing CYP1B1 mutations varies among different populations ranging from less than 10% to 100% [11,23-26]. In current study, CYP1B1 mutations were found in 20% (7/35) of the patients, which is comparable with the prevalence of CYP1B1 mutations in China (17.2%) [27], in Indonesia (33.3%) and in Europe (22.2%) [26]. Higher results are reported in the Indian (44%) and the Iranian population (70%) [28].
In the present study, 43% of patients harbouring CYP1B1 mutations showed family history of PCG, whereas another study reported that 87% of familial PCG cases are due to mutations in this gene [17]. Many studies said that CYP1B1 mutations are more common in populations where consanguinity is customary [29]. In current study, 100% of PCG patients harbouring CYP1B1 mutations had consanguineous parents. These results are in accordance with another study conducted in Saudi Arabia where among PCG patients, prevalence of CYP1B1 mutation ranged from 70% to 100% in consanguineous and inbred populations [30]. As Pakistani population is highly consanguineous, PCG segregating itself in recessive form is often seen in pediatric ophthalmic clinics. That’s why this study was planned to screen children affected by PCG and identify the underlying molecular defect. Previously, mutation in genes including CYP1B1 has been reported in Pakistani patients [31,32].
In the current study, sequencing analysis revealed one mutation R390H in seven patients, i.e. in 20% of the patients. These results are quite comparable with other studies that revealed 16% and 19.2% R390H mutations in CYP1B1 in PCG patients among Indian and Iranian population respectively [28,33]. Another study revealed p.R390H was the most common mutation in the Pakistani population accounting for 25% of (5/20) of the families with PCG [34].
In our study, all patients who harboured the c.1169 G>A mutation were homozygous, and one of these patients was unilaterally affected while rest were bilaterally affected by birth. These results are similar to findings of the study conducted in Iran and India that revealed all patients who harbour the c.1169 G>A mutation was homozygous showing clinical features and disease severity of varying degrees [28,31]. Thus, 1169 G>A has been reported to show variable phenotypes in various populations. Unknown environmental and genetic factors may be involved in the above mentioned variable clinical manifestations of CYP1B1 mutations.
The arginine (R) 390 residue is present in the conserved domain of alpha-helix K domain of CYP1B1 and thus is vital for the normal functioning of the CYPB1 protein [20]. The mutation c.1169 G>A (p.R390H) alters the side-chain physiognomies from linear guanidinium group to imidazole ring and disturbs the establishment of salt bridges on the ‘meander’ region [20].
Two missense sequence variants c.1294G>C and c.1358A>G carried by two and four patients respectively; in addition to the mutation mentioned above, were also observed in current research work. Both of these variations are previously reported and are considered as polymorphisms as not associated with the disease [25,28]. Besides a synonymous variant c.1347T>C was also found in the current study. However, this change in DNA nucleotide sequence does not result in an amino acid change (GAT>GAC Aspartic acid> Aspartic acid). This sequence variant was found in the homozygous state in 18 patients and 3 patients in heterozygous form. This CYP1B1 polymorphism variation is reported previously [25,28].
Our findings conclude that CYP1B1 mutation is one of the leading cause of PCG. Molecular characterization of c.1169 G>A (p.R390H) mutation using in-silico tools may help to understand the underlying pathology of the disease. These results will be beneficial and helpful in the diagnosis, treatment of PCG patients. The identification of the molecular cause of PCG will provide better ways of genetic counselling under or at reproductive age. Characterisation of CYP1B1 mutations may help to implement genetic analysis services for patients with PCG, and the genotype-phenotype correlation will aid in better management for the disease.
Declarations
Ethics approval and consent to participate
Before the start of the study, approval was obtained from the Advanced Studies & Research Board and Ethical Review Committee, University of Health Sciences, Lahore. Informed consent was received from the guardians of all the subjects who participated in the study, consistent with the tenets of the Declaration of Helsinki.
Availability of data and material
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Funding
The University of Health Sciences, Lahore, Pakistan, funded the entire project.
Authors' Contribution
MUK collected the samples of PCG patients, performed experimental work, interpreted the data, and prepared the manuscript. Bioinformatics analysis was performed by RR and AA, whereas SM and HK supervised the research work. All authors read and approved the final manuscript.
The authors declare that there is no conflict of interest regarding the publication of this paper.
Acknowledgement
We are thankful to the patients who took part in the study by providing their blood samples, clinical and family history. We are also grateful to the doctors of Layton Rahmatullah Benevolent Trust (LRBT), Lahore for co-operation during this study.
References![]()
- ourne RR, Flaxman SR, Braithwaite T, Cicinelli MV, Das A, et al. Magnitude, temporal trends, and projections of the global prevalence of blindness and distance and near vision impairment: a systematic review and meta-analysis. The Lancet Global Health, (2017); 5(9): e888-e897.
- Guo L, Salt TE, Luong V, Wood N, Cheung W, et al. Targeting amyloid-β in glaucoma treatment. Proceedings of the National Academy of Sciences, (2007); 104(33): 13444-13449.
- Vasiliou V, Gonzalez FJ. Role of CYP1B1 in glaucoma. Annu Rev Pharmacol Toxicol, (2008); 48333-358.
- Alex Kozak SGP, MD, MPH, K. David Epley, M.D., Robert A. Clark M.D. and Ryan.C.Plumb.AAO (2019) Glaucoma, Congenital Or Infantile. 22 October 2019 ed. USA: American Academy of Opthalmology.
- Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. British journal of ophthalmology, (2006); 90(3): 262-267.
- Kumar DM, Simpkins JW, Agarwal N. Estrogens and neuroprotection in retinal diseases. Molecular vision, (2008); 141480.
- Pang CP, Fan BJ, Canlas O, Wang DY, Dubois S, et al. A genome-wide scan maps a novel juvenile-onset primary open angle glaucoma locus to chromosome 5q. Mol Vis, (2006); 1285-92.
- Akarsu AN, Turacli ME, Aktan SG, Barsoum-Homsy M, Chevrette L, et al. A second locus (GLC3B) for primary congenital glaucoma (Buphthalmos) maps to the 1p36 region. Human molecular genetics, (1996); 5(8): 1199-1203.
- AlFadhli S, Behbehani A, Elshafey A, Abdelmoaty S, Al-Awadi S. Molecular and clinical evaluation of primary congenital glaucoma in Kuwait. American journal of ophthalmology, (2006); 141(3): 512-516.
- Papadopoulos M, Cable N, Rahi J, Khaw PT. The British infantile and childhood glaucoma (BIG) eye study. Investigative ophthalmology & visual science, (2007); 48(9): 4100-4106.
- Reddy A, Kaur K, Mandal AK, Panicker SG, Thomas R, et al. Mutation spectrum of the CYP1B1 gene in Indian primary congenital glaucoma patients. Mol Vis, (2004); 10696-702.
- Guo L, Moss SE, Alexander RA, Ali RR, Fitzke FW, et al. Retinal ganglion cell apoptosis in glaucoma is related to intraocular pressure and IOP-induced effects on extracellular matrix. Investigative ophthalmology & visual science, (2005); 46(1): 175-182.
- Stoilov I, Sarfarazi M. The third genetic locus (GLC3C) for primary congenital glaucoma (PCG) maps to chromosome 14q24. 3. Investigative ophthalmology & visual science, (2002); 43(13): 3015-3015.
- Firasat S, Riazuddin SA, Hejtmancik JF, Riazuddin S. Primary congenital glaucoma localizes to chromosome 14q24. 2-24.3 in two consanguineous Pakistani families. Molecular vision, (2008); 141659.
- Sarfarazi M, Akarsu NA, Hossain A, Turacli EM, Aktan GS, et al. Assignment of a locus (GLC3A) for primary congenital glaucoma (Buphthalmos) to 2p21 and evidence for genetic heterogeneity. Genomics, (1995); 30(2): 171-177.
- Ho CL, Walton DS. Primary congenital glaucoma: 2004 update. Journal of pediatric ophthalmology and strabismus, (2004); 41(5): 271-288.
- Nelson DR, Zeldin DC, Hoffman SM, Maltais LJ, Wain HM, et al. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics and Genomics, (2004); 14(1): 1-18.
- Chambers D, Wilson L, Maden M, Lumsden A. RALDH-independent generation of retinoic acid during vertebrate embryogenesis by CYP1B1. Development, (2007); 134(7): 1369-1383.
- Stoilov I, Jansson I, Sarfarazi M, Schenkman JB. Roles of cytochrome p450 in development. Drug metabolism and drug interactions, (2001); 18(1): 33-56.
- Stoilov I, Akarsu AN, Alozie I, Child A, Barsoum-Homsy M, et al. Sequence analysis and homology modeling suggest that primary congenital glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved core structures of cytochrome P4501B1. The American Journal of human genetics, (1998); 62(3): 573-584
- Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, et al. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic acids research, (1989); 17(20): 8390.
- Kaul H, Riazuddin SA, Shahid M, Kousar S, Butt NH, et al. Autosomal recessive congenital cataract linked to EPHA2 in a consanguineous Pakistani family. Molecular vision, (2010); 16511.
- Bejjani BA, Stockton DW, Lewis RA, Tomey KF, Dueker DK, et al. Multiple CYP1B1 mutations and incomplete penetrance in an inbred population segregating primary congenital glaucoma suggest frequent de novo events and a dominant modifier locus. Human molecular genetics, (2000); 9(3): 367-374.
- Mashima Y, Suzuki Y, Sergeev Y, Ohtake Y, Tanino T, et al. Novel cytochrome P4501B1 (CYP1B1) gene mutations in Japanese patients with primary congenital glaucoma. Investigative ophthalmology & visual science, (2001); 42(10): 2211-2216.
- Colomb E, Kaplan J, Garchon HJ. Novel cytochrome P450 1B1 (CYP1B1) mutations in patients with primary congenital glaucoma in France. Human mutation, (2003); 22(6): 496-496.
- Sitorus R, Ardjo S, Lorenz B, Preising M. CYP1B1 gene analysis in primary congenital glaucoma in Indonesian and European patients. Journal of medical genetics, (2003); 40(1): e9-e9.
- Chen Y JD, Yu L, Katz B, Zhang K, Wan B, Sun X. CYP1B1 and MYOC mutations in 116 Chinese patients with primary congenital glaucoma. Archives of ophthalmology (2008); 126(10): 05.
- Chitsazian F, Tusi BK, Elahi E, Saroei HA, Sanati MH, et al. CYP1B1 mutation profile of Iranian primary congenital glaucoma patients and associated haplotypes. The Journal of Molecular Diagnostics, (2007); 9(3): 382-393.
- Vogt G, Horváth‐Puhó E, Czeizel AE. A population‐based case‐control study of isolated primary congenital glaucoma. American Journal of Medical Genetics Part A, (2006); 140(11): 1148-1155.
- Abu-Amero KK, Osman EA, Mousa A, Wheeler J, Whigham B, et al. Screening of CYP1B1 and LTBP2 genes in Saudi families with primary congenital glaucoma: genotype-phenotype correlation. Molecular vision, (2011); 172911.
- Firasat S, Kaul H, Ashfaq UA, Idrees S. In silico analysis of five missense mutations in CYP1B1 gene in Pakistani families affected with primary congenital glaucoma. International ophthalmology, (2017); 1-8.
- Rauf B, Irum B, Kabir F, Firasat S, Naeem MA, et al. A spectrum of CYP1B1 mutations associated with primary congenital glaucoma in families of Pakistani descent. Human genome variation, (2016); 316021.
- Tanwar M, Dada T, Sihota R, Das TK, Yadav U, et al. Mutation spectrum of CYP1B1 in North Indian congenital glaucoma patients. Molecular vision, (2009); 151200.
- Sheikh SA, Waryah AM, Narsani AK, Shaikh H, Gilal IA, et al. Mutational spectrum of the CYP1B1 gene in Pakistani patients with primary congenital glaucoma: novel variants and genotype-phenotype correlations. Molecular vision, (2014); 20991.
![]()
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0