

Review Article
The genetics associated with Primary Congenital Glaucoma
Ali Imran1, Muhammad Umer Khan1*, Umer Nasir1, Qasim Qayyum1, Rubab Hector1, Raima Rehman1, Atif Amin Baig 2
Adv. life sci., vol. 7, no. 2, pp. 106-112, February 2020
*- Corresponding Author: Muhammad Umer Khan (Email: umer.khan685@gmail.com)
Authors' Affiliations
2. University Sultan Zainul Abdin – Malaysia
Abstract![]()
Introduction
Methods
Discussion
Conclusion
References
Abstract
Glaucoma is a progressive optic neuropathy; increased intraocular pressure (IOP) is a modifiable risk factor for primary congenital glaucoma (PCG). Increase IOP causes retinal and optic nerve compression and leads to gradual and irreversible loss of eyesight if left untreated. It is the second most leading cause of blindness. PCG mainly affects children up to the age of three years, and symptoms include epiphora, photalgia, swollen eyes, opaque corneas, blepharospasm, rupture in the retina and ocular nerve damage due to IOP. Early detection, management, and treatment are the keys to preventing vision loss from glaucoma. Many mutations have been discovered in Cytochrome P450 1B1 (CYP1B1) gene to be responsible for causing PCG, and there are still a lot of mutations to be discovered. In this review, we will discuss the genetic aspects of PCG and the most frequent mutations responsible for PCG in Pakistani children. PCG can be handled by decreasing IOP either by medication or by surgery. Genetic counselling plays a significant role in the establishment of proper management of PCG.
Keywords: Primary Congenital Glaucoma; IOP; Cyp1b1; Mutations
Introduction![]()
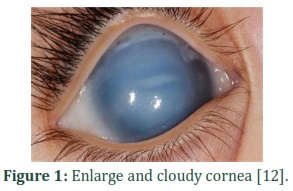
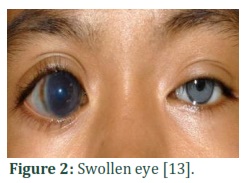
Glaucoma (pronounced: glawko’me) [1], which is originated from Greek word ‘glaukos’ (denoting blue-greenish blink) [2] is an accumulation of various diseases associated with optic nerve damage [3] in which huge waste of retinal ganglion cells (RGC) occurs [4,5] and carbon copy shape of visual field and vision is dissipated [6].The main clinical mark of glaucoma is the raise in aqueous humor build up in the anterior chamber [7]. The inheritance is mainly autosomal recessive, while a dominant transmission way has also been illustrated [8]. Glaucoma has casual symptoms and consists of rupture in retina, light susceptibility, eye scrub, and irritation. Increased intraocular pressure causes enlarge, cloudy, and opaque cornea (Fig.1), swollen eyes (Fig.2) and ocular nerve damage [4,9]. Glaucoma is the most critical cause of bilateral blindness around the world. The approximated occurrence of glaucoma is 64.3 million, of whom 8.4 million people are bilaterally blind [10]. This frequency is expected to increase with a shocking rate to 76 million in 2020 and 111.8 million in 2040 [11].
Methods![]()
Literature search and selection criteria:
Different search engines such as PubMed, Google Web, Science Direct, Google Scholar and Research gate were used to retrieve the data for review write up. Glaucoma, CYP1B1, PCG, IOP were used as keywords for searching the related data. In this study, 70 peer-reviewed research articles were selected. Manuscripts with other types of glaucoma were excluded to write up this review.
Types of Glaucoma:
Commonly, glaucoma has following three subtypes according to the age of infancy, cause of disease, and structure of the anterior chamber, primary open-angle glaucoma (POAG), primary angle-closure glaucoma (PACG) and primary congenital glaucoma (PCG) [14]. The Major form of glaucoma is POAG, holding for 80% of glaucomatous diseases, while PCG is accepted as another critical form of glaucoma in infants, regardless of its low occurrence [14,15].
Primary congenital glaucoma:
Primary congenital glaucoma happens before the age of three years without visible structural error of eye[16]. It is developed by increased intraocular pressure (IOP) [17]. Aqueous fluid produced in the ciliary body is filled in the frontal part of the eye and leaves the optic across trabecular meshwork which is a porous tissue present in the point from where cornea penetrates the iris. Deformity in shape or form of the iridocorneal angle may limit the drain of aqueous humour and ultimately increase IOP,it is a firm cause for the development of glaucoma[18] and constitutes up to 18% of childhood blindness[19,20].At birth, 50% of patients reveal symptoms, and at the age of 1 year 80% of patients are confirmed with PCG; of these, 65% of patients are male and 70% of patients having symptoms of bilateral blindness [21].
Genetics of PCG:
In the early 1990s, the genetic heterogeneity studies for PCG were begun. In 1995 the first locus for PCG on 2p21 chromosomal location was mapped [22]. Loci of five chromosomes, GLC3A (chromosome 2p21), GLC3B (chromosome 1p36.2-p36.1), GLC3C (chromosome 14q24.3), GLC3D (chromosome 14q24.2-q24.3), and GLC3E (chromosome 9p21.2) are directly related with the disease [23]. However, in the development of PCG, only three genes are involved: Cytochrome P450 1B1 (CYP1B1) which islocated in the GLC3A locus; Latent Transforming growth factor-beta binding protein 2 (LTBP2) situated in the GLC3D; and Tunica interna endothelial cell kinase (TEK) is located in the GLC3E. In this disease role of protein encoded by three genes is still uncertain [23-25]. CYP1B1 or LTBP2 cause PCG in an autosomal recessive manner. Heterozygous pathogenic variants in TEK cause PCG in an autosomal dominant manner [17].
In autosomal recessive inheritance at insemination, each sibling of a diseased parent has a 25% possibility of being affected, 50% possibility of being a non-symptomatic carrier, and 25% possibility of being healthy. Heterozygotes (carriers) show no symptoms; carrier testing of family members is viable if the pathogenic form in the family is recognised [17]. In autosomal dominant inheritance, there is 50% probability in offspring of TEK-related PCG individual to inherit pathogenic form. Early diagnosis of at-risk pregnancy is feasible if mutant variants of PCG are detected in a family [17].
Epidemiology:
In 1842, glaucoma was first reported by Benedict when he discovered glaucoma in two sisters [26]. PCG is more frequent in the Middle East, including Saudi Arabia [27]. PCG is ten times more common in some ethnic groups, where consanguineous relationships, especially cousin-marriages are common [28,29].The incidence of PCG is high in Slovakia and Saudi Arabia, where 1 in 1,250 and 1 in 2500 individuals are affected, respectively [30].PCG has a variable prevalence in disparate and ethnic groups. The prevalence of PCG in Western countries, like Britain, Ireland and the USA is 1 per 10 to 20,000 live births [3,31-34]. PCG has the lowest incidence in eastern and northern province of Saudi Arabia,11.1% and 9% respectively[35]. The Middle East has 64.8% and the Maghreb has 54.4% of CYP1B1 mutations. The percentage of these mutations in Europe, Asia and in the United States is 34.7%, 21.3%, and 14.9% respectively. Founder mutations have been discovered in different geographical areas. For example, the most frequently reported variants, p.Arg390His, p.Gly61Glu, p.Val320Leu, p.E387Lys and p.Gly61Glu were observed in Pakistan, Iran/Saudi Arabia, Morocco, Vietnam/South Korea, Europe, and in Lebanon. These discovered variants in seven different countries were quite similar to mutations in Morocco. These indications tell us about geographical distribution as well as genetic differences of PCG mainly involve with CYP1B1 gene variation. In the hereditary screening of patients with PCG, the first step should examine for founder and typical mutations [36]. PCG appears within a few months of life if the family has a high prevalence of PCG. In India, Asia and Saudi Arab, the signified age for having PCG, is from 3 to 4 months and for Western countries it is up to11 months [35,37].
Cytochrome P450 1B1 (CYP1B1):
CYP1B1 belongs to the family of cytochrome P450. This oxidase enzyme is bound in the membrane and during development play extensive functions in metabolism of hormones and involve in different metabolic activity [38]. In liver cytochromes are most expressed and in colon,lung,eye and kidney CYP1B1 is more proliferate [39]. The mechanism of PCG caused by a mutation in CYP1B1 needs to be further explored. Trabecular meshwork and ciliary body with CYP1B1 mutations have high intraocular pressure in the eye [24,40]. Current observation suggests that for the growth and functioning of the trabecular meshwork, CYP1B1 might be necessary[41,42] and that may be a mutation in CYP1B1 gene change function of trabecular meshwork and impair the regulation of intraocular pressure, optic nerve destruction, and eventually PCG [16].
CYP1B1 gene was sequestered and mapped on chromosomal position 2p21-22 by Tang et al [43]. There are three exons in CYP1B1 gene of human (start with exon II ORF) and two introns and is 8.5 Kb in length. There are 371, 1,044 and 3,707 base pairs respectively in all three exon lengths. In contrast, the length of two introns in base pair is 390 and 3032. These two introns start with sequence of GT and end with sequence of AG.Pyrimidine is abundant in the upstream region of introns, in 2nd exon, coding part of CYP1B1 gene is initiated from 5’ end and finish in terminal exon. Five hundred forty-three amino acids are translation outcome of CYP1B1 gene (relate to 1,629 bases). Carboxyl terminal part of CYP1B1 gene is most preserve, recommended a vital function of this part. For normal functioning of cytochrome P450 enzyme, carboxyl-terminal has conserved core-like structures in heme binding part. By using DNA probes against three exons of CYP1B1 gene in southern analysis it is confirmed that it is a single-copy gene. TATA box is missing in the promoter part of the CYP1B1 gene and consist of nine TCDD reactive enhancer regions and placed in 2.5 kb upstream [44].
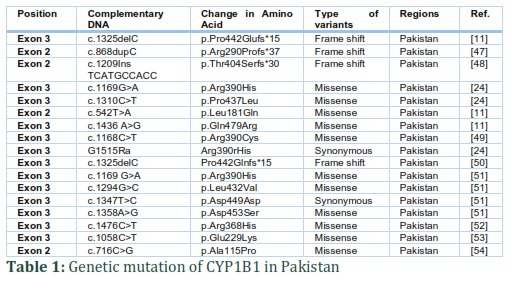
CYP1B1 is a component of cytochrome P450, which is mono-oxygenase protein and belongs to superfamily of enzyme and triggers many reactions involving metabolism of drugs, and formation of steroids, cholesterol and lipids. CYP1B1 binds to endoplasmic reticulum and involved in metabolism of procarcinogens (PAHs compounds) [17]. In the Human Gene Mutation database around 240 pathogenic variants of CYP1B1 are listed [11] along with missense and nonsense variants, small deletions/insertions/duplications, and exon and whole-gene deletions [17]. Many studies have carried out by using computer simulation and in laboratory to identify the effect of mutation in CYP1B1 on composition as well as working of protein. Jansson et al. in laboratory conducted the impact of CYP1B1 mutation on the reliability and working of protein. He investigates the effect of two missense variations (p.Ala469Thr and p.Gly61Glu) on enzymatic role and reliability of CYP1B1. He discovered the variant protein p.Gly61Glu had missed 60% reliability, although p.Ala469Thr preserves almost 80% reliability over wild type. Enzymatic technique was used to know the impact of variant on protein working. When contrast to wild type protein, the result establish low metabolic function (50% – 70%) for whole substrates [17]. In 542 PCG patients,147 different mutations are detected in CYP1B1 gene around the world [45]. These contain insertions, deletions, nonsense, missense, frameshift and also truncating mutations and variation in noncoding part of exon I. The most critical phenotype is related to the frameshift mutations [46]. Genetic mutation of CYP1B1 in Pakistani population is shown in table 1.
Latent transforming growth factor-beta binding protein 2 (LTBP2):
LTBP2 mutations are infrequent in PCG diseases. In Pakistan and Iran, most of the cases that were reported having cousin marriages in the family [55,56]. LTBP2 form protein outside the cell with the role being considered in cell attachment [57,58] and elastin microfibril assembly [59-61]. LTBP2 is mainly present in those tissues which are rich in fibres like arteries and lungs [53]. LTBP2 is majorly found in eye tissues, and those are involved majorly in maintenance of intraocular pressure and biology of glaucoma, as well as trabecular meshwork and ciliarybody.In addition, LTBP2 is mandatory for growth of frontal chamber as well as ciliaryzonules [55,62]. As a result of Variation in LTBP2 hereditary diseases of orbital structure develop, which raised intraocular pressure and cause PCG [16]. LTBP2 transcript NM_000428.2 comprises 36 exons [17]. The transformed protein NP_000419.1, constituting 1821 amino acids, from the group of latent transforming growth factor (TGF)-beta binding proteins (LTBP).This protein is extracellular ground substance with multidimensional shape, which is the biggest constituent of the LTBP group. It has so far been recommended that the protein may have varying tasks, as a part of the TGF-beta silent compound, as a composed part of microfibrils, and as an intermediate of cell [17].
In the Human Gene Mutation database,around 26 pathogenic variants of LTBP2 are listed [11]. Pathogenic mutations may damage shape of protein as well as upset functions of protein, also change in both fibrillin 1 and fibulin 5 binding [17].
Tunica interna endothelial cell kinase (TEK):
TEK mediate the process of blood vessel formation, mainly present in endothelium as well as in lymphatic endothelia [63,64]. It Plays vital function in glaucoma; this is probably said TEK variants lead to change in the growth of orbital shape which is compulsory for aqueous drainage tract as well as maintenance of intraocular pressure and leading to congenital glaucoma [16].TEK has different copies of mutation; the largest from them is NM_000459.4 that is 4.7kb long and contains 23 exons [17]. TEK form tyrosine-protein kinase which plays role in cell-surface receptor, the angiopoietin-1 receptor. It is present totally in endothelial cells [17]. In the Human Gene Mutation database, 10 disease-causing mutations are recorded, plus missense, nonsense, and dividing mutations, little removal & addition lead to frameshift mutations [17].Pathogenic mutation in TEK outcome in venous deformity in nonocular tissues, whereas dysfunction mutation upset anterior chamber vascular development and result in primary congenital glaucoma [17].
Diagnosis:
The structural and clinical changes due to glaucoma are permanent.Therefore; PCG should be detected timely to save visual loss. Early diagnosis can be made by observing optic nerve structure through imaging devices and assess function of optic nerve by using perimetry [65].
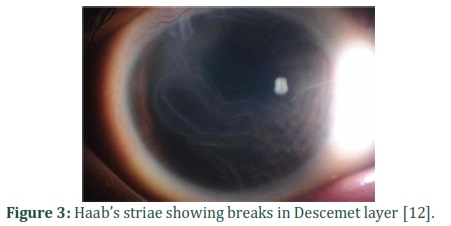
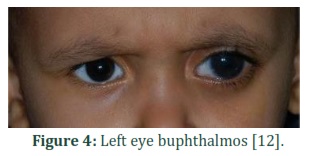
Glaucoma can be symmetrical or asymmetrical, unilateral or bilateral. The condition can be observed at birth or after a few months of birth. In situations where PCG is suspicious, emergency eye examination is required under general anaesthesia or sedation to measure coronary diameter and IOP [66,67]. In PCG, diameter of cornea increases abnormally, which is up to 10mm at birth. The visibility of cornea is changed due to corneal oedema, which intensifies rapidly[67]. In usually, intraocular pressure in a child often is about 12.02 mm Hg.Increased in IOP from 21 mm Hg (mercury) in eyes as checked by I-care tonometry™, applanation tonometry, and pneumotonometry on different occasion is evaluated as abnormal. The recognition of PCG is depended on hospital checkup which involves raised IOP in children typically earlier than one year of age, expansion of the globe, increased corneal diameter, dull corneas, breaks in Descemet’s membrane (Haab’striae) (Fig.3), buphthalmos (Fig.4) and anomalously deep anterior chamber. The detection of PCG is not difficult when a child has heavy hallmarks. If clinical hallmarks are not revealed then it is tough to diagnose PCG, mainly if PCG is bilateral. However, early detection is vital to initiate rapid care and secure final effective results. The situation is primarily identified at initial months after birth[66,67].Molecular testing proceeded towarduni-gene checking, use of a multigene panel, as well as complete genomic checking can help in diagnosis [17].
Treatment and Management:
Undiagnosed and untreated glaucoma can be a source of irreversible blindness. Medications, laser therapy and surgical incision are treatment options for glaucoma patients. There are certain risks and advantages of all types of treatment. Therefore, treatment should be carefully selected to minimise the adverse effects while maximising benefits of treatment. Usual first-line treatment of glaucoma routinely starts with the use of topical selective or non-selective β-blockers or topical compounds like latanoprost, travoprost, and bimatoprost. Topical carbon anhydrase inhibitors and α-agonists are considered second-line treatment options. Third-line drugs of choice include Para sympathomimetic agents, most frequently pilocarpine. If medications fail than other methods, laser trabeculoplasty and Incisional surgery can be used to lower IOP [68].
The main surgical options for PCG are angle surgery and drainage surgery.Surgery is best option [69]. If the eye can improve optic visualisation, then it is necessary to stop more optical collapse. In contrast, if delay identification occurs than a high amount of permanent destruction had previously happened and in these cases, treatment is intended to maintain present optic state and help to stop upcoming collapse [12]. Due to remarkable anatomical abnormality of frontal drainage angle there are some limitations in treatment of PCG by decreasing IOP. In PCG patients reduction in IOP is less than 10% effective [70].
If surgery is unsuccessful than medicine is given to normalise intraocular pressure and routine treatment is accomplished for refractive errors and amblyopia [17]. Cyclodestruction and glaucoma drainage devices can help to maintain intraocular pressure. Glaucoma drainage devices usage data has release 28%-49% decrease in mean intraocular pressure and behind one year of surgical operation achievement rate is 63%-97% [71-73].
Before surgery, discontinuation of medications such as Phospholine Iodide (echothiophate) is essential, mainly if Succinylcholine is used to prevent cessation of breathing. It can help to avoid from secondary complications Alpha-2 agonists should not be taken because of risk for cessation of breathing and decrease in heart rate. Lifetime follow up is essential to control intraocular pressure to protect residual eyesight and stop more loss of vision. Monitoring depends upon control of IOP and extremity of disease. When IOP is control and the child is visually re-establish than every three months follow up is usually done to maintain the IOP at normal level and it dependson the age of individual and extremity of optic nerve harm. Visual field testing, as well as optic nerve photography are included in follow-up tests. Sedation and anaesthesia are essential for the proper ophthalmic observation in newborns and uncooperative youngsters. This procedure is difficult for the patient, the family and the treating consultant [17]. Proper testing of newborn babies in early life may diagnose PCG in their childhood and they can be treated earlier and avoid from routinely checkup under anaesthesia. If the pathogenic variants have been detected than only genetic testing is suitable in sibling of a diseased person. In an affected family member, if the PCG related pathogenic variants have not been recognised, then member screening includes IOP evaluations under anaesthesia [17].
Figures & Tables
Conclusion![]()
Primary congenital glaucoma is one of the significant vexing eye alarming problems, which can cause permanent loss of vision if left untreated. PCG represents a common disorder where genetic screening can be very beneficial for ethnic and prenatal diagnoses of at-risk individuals. It has high prevalence in population where consanguinity is common. Factors that can increase the burden of disease are limited resources, delayed presentation, consanguineous relationships, and limited follow-up. This review article started to explore genetic aspects of PCG, especially CYP1B1, LTBP2, TEK and diagnosis and management of PCG to gain a deeper understanding of PCG. Early and precise diagnosis of PCG is essential for predictive testing and early intervention to minimise the effects of visual impairment and eventually blindness. Risk of PCG can be reduced by avoiding consanguinity. Efforts to improve and expand PCG awareness programs must continue in the developing world, and give comprehensive treatment to PCG patients.
Authors' Contribution
RH and QQ collected all the relevant manuscripts, AI and UN prepared the initial draft of the manuscript. MUK and RR finalized the manuscript, whereas MUK and AAB technically reviewed the manuscript. All authors read and approved the final manuscript.
The authors declare that there is no conflict of interest regarding the publication of this paper.
References![]()
- Pemberton SG, Frey RW. Trace fossil nomenclature and the Planolites-Palaeophycus dilemma. Journal of Paleontology, (1982); 843-881.
- Dictionary OE. Oxford english dictionary. Simpson, JA & Weiner, ESC, (1989).
- Sarfarazi M, Stoilov I, Schenkman J. Genetics and biochemistry of primary congenital glaucoma. Ophthalmology clinics of North America, (2003); 16(4): 543-554.
- deLuise VP, Anderson DR. Primary infantile glaucoma (congenital glaucoma). Survey of Ophthalmology, (1983); 28(1): 1-19.
- Quigley HA. Open-angle glaucoma. New England Journal of Medicine, (1993); 328(15): 1097-1106.
- Weinreb RN, Khaw PT. Primary open-angle glaucoma. The Lancet, (2004); 363(9422): 1711-1720.
- Anderson J (1939) Aetiology: Hydrophthalmia or Congenital Glaucoma. London, England: Cambridge University Press.
- Hewitt A, Mackinnon J, Elder J, Giubilato A, Craig J, et al. Familial transmission patterns of infantile glaucoma in Australia. Investigative Ophthalmology & Visual Science, (2005); 46(13): 3207-3207.
- Ko F, Papadopoulos M, Khaw PT (2015) Primary congenital glaucoma. Progress in brain research: Elsevier. pp. 177-189.
- Rulli E, Quaranta L, Riva I, Poli D, Hollander L, et al. Visual field loss and vision-related quality of life in the Italian Primary Open Angle Glaucoma Study. Scientific reports, (2018); 8(1): 619.
- Rashid M, Yousaf S, Sheikh SA, Sajid Z, Shabbir AS, et al. Identities and frequencies of variants in CYP1B1 causing primary congenital glaucoma in Pakistan. Molecular vision, (2019); 25: 144-154.
- Badawi AH, Al-Muhaylib AA, Al Owaifeer AM, Al-Essa RS, Al-Shahwan SA. Primary congenital glaucoma: An updated review. Saudi Journal of Ophthalmology, (2019); 33(4): 382-388.
- Fan BJ, Wiggs JL. Glaucoma: genes, phenotypes, and new directions for therapy. The Journal of clinical investigation, (2010);120(9):3064-72.
- de Alencar Gomes H, de Souza Moreira B, Sampaio RF, Furtado SRC, Cronemberger S, et al. Gait parameters, functional mobility and fall risk in individuals with early to moderate primary open angle glaucoma: a cross-sectional study. Brazilian Journal of Physical Therapy, (2018); 22(5): 376-382.
- Yang Y, Zhang L, Li S, Zhu X, Sundaresan P. Candidate gene analysis identifies mutations in CYP1B1 and LTBP2 in Indian families with primary congenital glaucoma. Genetic Testing and Molecular Biomarkers, (2017); 21(4): 252-258.
- Lewis CJ, Hedberg-Buenz A, DeLuca AP, Stone EM, Alward WL, et al. Primary congenital and developmental glaucomas. Human Molecular Genetics, (2017); 26(R1): R28-R36.
- Abu-Amero KK, Edward DP (2017) Primary congenital glaucoma. GeneReviews®[Internet]: University of Washington, Seattle.
- Jünemann A, Hohberger B, Rech J, Sheriff A, Fu Q, et al. Agonistic autoantibodies to the β2-adrenergic receptor involved in the pathogenesis of open-angle glaucoma. Frontiers in Immunology, (2018); 9: 145.
- Kwon YH, Fingert JH, Kuehn MH, Alward WL. Primary open-angle glaucoma. New England Journal of Medicine, (2009); 360(11): 1113-1124.
- Gilbert CE, Canovas R, de Canovas RK, Foster A. Causes of blindness and severe visual impairment in children in Chile. Developmental Medicine & Child Neurology, (1994); 36(4): 326-333.
- Zhao J. Prevention of blindness is still the great challenge faced by Chinese ophthalmology. Chinese Journal of Ophthalmology, (2009); 45(9): 769-771.
- Sarfarazi M, Akarsu NA, Hossain A, Turacli EM, Aktan GS, et al. Assignment of a locus (GLC3A) for primary congenital glaucoma (Buphthalmos) to 2p21 and evidence for genetic heterogeneity. Genomics, (1995); 30(2): 171-177.
- Chen Y, Jiang D, Yu L, Katz B, Zhang K, et al. CYP1B1 and MYOC mutations in 116 Chinese patients with primary congenital glaucoma. Archives of Ophthalmology, (2008); 126(10): 1443-1447.
- Stoilov I, Akarsu AN, Alozie I, Child A, Barsoum-Homsy M, et al. Sequence analysis and homology modeling suggest that primary congenital glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved core structures of cytochrome P4501B1. The American Journal of human genetics, (1998); 62(3): 573-584.
- López-Garrido M-P, Medina-Trillo C, Morales-Fernandez L, Garcia-Feijoo J, Martínez-de-la-Casa JM, et al. Null CYP1B1 genotypes in primary congenital and nondominant juvenile glaucoma. Ophthalmology, (2013); 120(4): 716-723.
- Benedict TWG Abhandlungen aus dem Gebiete der Augenheilkunde. 1842; 1; Freund.
- Jaafar MS. Care of the infantile glaucoma patient. Ophthalmology annual, (1988); 715-37.
- Papadopoulos M, Cable N, Rahi J, Khaw PT. The British infantile and childhood glaucoma (BIG) eye study. Investigative Ophthalmology & Visual Science, (2007); 48(9): 4100-4106.
- Qayyum M, Zia WT, Khan MU. An Insight to Primary Congenital Glaucoma. Critical Review in Eukaryotic Gene Expression, (2020); 30(1); 39-43.
- Bejjani BA, Lewis RA, Tomey KF, Anderson KL, Dueker DK, et al. Mutations in CYP1B1, the gene for cytochrome P4501B1, are the predominant cause of primary congenital glaucoma in Saudi Arabia. The American Journal of Human Genetics, (1998); 62(2): 325-333.
- Genĉík A. Epidemiology and genetics of primary congenital glaucoma in Slovakia. Description of a form of primary congenital glaucoma in gypsies with autosomal-recessive inheritance and complete penetrance. Developments in Ophthalmology, (1989); 1676-115.
- Genčík A, Genčíkova A, Ferak V. Population genetical aspects of primary congenital glaucoma. I. Incidence, prevalence, gene frequency, and age of onset. Human Genetics, (1982); 61(3): 193-197.
- MacKinnon JR, Giubilato A, Elder JE, Craig JE, Mackey DA. Primary infantile glaucoma in an Australian population. Clinical & Experimental Ophthalmology, (2004); 32(1): 14-18.
- Tamçelik N, Atalay E, Bolukbasi S, Çapar O, Ozkok A. Demographic features of subjects with congenital glaucoma. Indian Journal of Ophthalmology, (2014); 62(5): 565.
- Alanazi FF, Song JC, Mousa A, Morales J, Al Shahwan S, et al. Primary and secondary congenital glaucoma: baseline features from a registry at King Khaled Eye Specialist Hospital, Riyadh, Saudi Arabia. American Journal of Ophthalmology, (2013); 155(5): 882-889. e881.
- Chouiter L, Nadifi S. Analysis of CYP1B1 gene mutations in patients with primary congenital glaucoma. Journal of Pediatric Genetics, (2017); 6(04): 205-214.
- Fung DS, Roensch MA, Kooner KS, Cavanagh HD, Whitson JT. Epidemiology and characteristics of childhood glaucoma: results from the Dallas Glaucoma Registry. Clinical Ophthalmology (Auckland, NZ), (2013);7: 1739-46.
- Stoilov I, Jansson I, Sarfarazi M, Schenkman JB. Roles of cytochrome p450 in development. Drug Metabolism and Drug Interactions, (2001); 18(1): 33-56.
- Li F, Zhu W, Gonzalez FJ. Potential role of CYP1B1 in the development and treatment of metabolic diseases. Pharmacology & Therapeutics, (2017); 178: 18-30.
- Stoilov I, Akarsu AN, Sarfarazi M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Human Molecular Genetics, (1997); 6(4): 641-647.
- Zhao Y, Wang S, Sorenson CM, Teixeira L, Dubielzig RR, et al. Cyp1b1 mediates periostin regulation of trabecular meshwork development by suppression of oxidative stress. Molecular and Cellular Biology, (2013); 33(21): 4225-4240.
- Mookherjee S, Acharya M, Banerjee D, Bhattacharjee A, Ray K. Molecular basis for involvement of CYP1B1 in MYOC upregulation and its potential implication in glaucoma pathogenesis. PloS one, (2012); 7(9): e45077.
- Tang YM, Wo Y-YP, Stewart J, Hawkins AL, Griffin CA, et al. Isolation and characterization of the human cytochrome P450 CYP1B1 gene. Journal of Biological Chemistry, (1996); 271(45): 28324-28330.
- Faiq M, Sharma R, Dada R, Mohanty K, Saluja D, et al. Genetic, biochemical and clinical insights into primary congenital glaucoma. Journal of Current Glaucoma Practice, (2013); 7(2): 66-84.
- Li N, Zhou Y, Du L, Wei M, Chen X. Overview of Cytochrome P450 1B1 gene mutations in patients with primary congenital glaucoma. Experimental Eye Research, (2011); 93(5): 572-579.
- Panicker SG, Mandal AK, Reddy AB, Gothwal VK, Hasnain SE. Correlations of genotype with phenotype in Indian patients with primary congenital glaucoma. Investigative Ophthalmology & Visual Science, (2004); 45(4): 1149-1156.
- Prokudin I, Simons C, Grigg JR, Storen R, Kumar V, et al. Exome sequencing in developmental eye disease leads to identification of causal variants in GJA8, CRYGC, PAX6 and CYP1B1. European Journal of Human Genetics, (2014); 22(7): 907-15.
- Kelberman D, Islam L, Jacques TS, Russell-Eggitt I, Bitner-Glindzicz M, et al. CYP1B1-Related Anterior Segment Developmental Anomalies: Novel Mutations for Infantile Glaucoma and Von Hippel's Ulcer Revisited. Ophthalmology, (2011); 118(9): 1865-1873.
- Curry SM, Daou AG, Hermanns P, Molinari A, Lewis RA, et al. Cytochrome P4501B1 mutations cause only part of primary congenital glaucoma in Ecuador. Ophthalmic Genetics, (2004); 25(1): 3-9.
- Reis LM, Tyler RC, Weh E, Hendee KE, Kariminejad A, et al. Analysis of CYP1B1 in pediatric and adult glaucoma and other ocular phenotypes. Molecular vision, (2016); 22: 1229-1238.
- Khan MU, Rehman R, Kaul H, Mahmood S, Ammar A. Mutational analysis of CYP1B1 gene in Pakistani pediatric patients affected with Primary Congenital Glaucoma. Advancements in Life Sciences, (2019); 7(1): 32-37.
- Rauf B, Irum B, Kabir F, Firasat S, Naeem MA, et al. A spectrum of CYP1B1 mutations associated with primary congenital glaucoma in families of Pakistani descent. Human Genome Variation, (2016); 3:16021.
- Firasat S, Kaul H, Ashfaq UA, Idrees S. In silico analysis of five missense mutations in CYP1B1 gene in Pakistani families affected with primary congenital glaucoma. International Ophthalmology, (2018); 38(2): 807-814.
- Sheikh SA, Waryah AM, Narsani AK, Shaikh H, Gilal IA, et al. Mutational spectrum of the CYP1B1 gene in Pakistani patients with primary congenital glaucoma: novel variants and genotype-phenotype correlations. Molecular vision, (2014); 20991.
- Ali M, McKibbin M, Booth A, Parry DA, Jain P, et al. Null mutations in LTBP2 cause primary congenital glaucoma. The American Journal of Human Genetics, (2009); 84(5): 664-671.
- Narooie-Nejad M, Paylakhi SH, Shojaee S, Fazlali Z, Rezaei Kanavi M, et al. Loss of function mutations in the gene encoding latent transforming growth factor beta binding protein 2, LTBP2, cause primary congenital glaucoma. Human Molecular Genetics, (2009); 18(20): 3969-3977.
- Vehviläinen P, Hyytiäinen M, Keski-Oja J. Latent transforming growth factor-β-binding protein 2 is an adhesion protein for melanoma cells. Journal of Biological Chemistry, (2003); 278(27): 24705-24713.
- Hyytiäinen M, Keski-Oja J. Latent TGF-β binding protein LTBP-2 decreases fibroblast adhesion to fibronectin. The Journal of Cell Biology, (2003); 163(6): 1363-1374.
- Hirai M, Ohbayashi T, Horiguchi M, Okawa K, Hagiwara A, et al. Fibulin-5/DANCE has an elastogenic organizer activity that is abrogated by proteolytic cleavage in vivo. J Cell Biol, (2007); 176(7): 1061-1071.
- Fujikawa Y, Yoshida H, Inoue T, Ohbayashi T, Noda K, et al. Latent TGF-β binding protein 2 and 4 have essential overlapping functions in microfibril development. Scientific reports, (2017); 7: 43714.
- Hirani R, Hanssen E, Gibson MA. LTBP-2 specifically interacts with the amino-terminal region of fibrillin-1 and competes with LTBP-1 for binding to this microfibrillar protein. Matrix Biology, (2007); 26(4): 213-223.
- Inoue T, Ohbayashi T, Fujikawa Y, Yoshida H, Akama TO, et al. Latent TGF-β binding protein-2 is essential for the development of ciliary zonule microfibrils. Human Molecular Genetics, (2014); 23(21): 5672-5682.
- Kizhatil K, Ryan M, Marchant JK, Henrich S, John SW. Schlemm's canal is a unique vessel with a combination of blood vascular and lymphatic phenotypes that forms by a novel developmental process. PLoS Biology, (2014); 12(7): e1001912.
- Park D-Y, Lee J, Park I, Choi D, Lee S, et al. Lymphatic regulator PROX1 determines Schlemm’s canal integrity and identity. The Journal of Clinical Investigation, (2014); 124(9): 3960-3974.
- Sung KR, Kim JS, Wollstein G, Folio L, Kook MS, et al. Imaging of the retinal nerve fibre layer with spectral domain optical coherence tomography for glaucoma diagnosis. British Journal of Ophthalmology, (2011); 95(7): 909-914.
- Dietlein TS, Jacobi PC, Krieglstein GK. Assessment of diagnostic criteria in management of infantile glaucoma. International ophthalmology, (1996); 20(1-3): 21-27.
- Beck AD. Diagnosis and management of pediatric glaucoma. Ophthalmology Clinics of North America, (2001); 14(3): 501-512.
- Lee DA, Higginbotham EJ. Glaucoma and its treatment: a review. American Journal of Health-System Pharmacy, (2005); 62(7): 691-699.
- Chan JYY, Choy BN, Ng AL, Shum JW. Review on the management of primary congenital glaucoma. Journal of Current Glaucoma Practice, (2015); 9(3): 92-99.
- Turaçh ME, Aktan G, Idil A. Medical and surgical aspects of congenital glaucoma. Acta Ophthalmologica Scandinavica, (1995); 73(3): 261-263.
- Al Faran MF, Tomey KF, Al Mutlaq FA. Cyclocryotherapy in selected cases of congenital glaucoma. Ophthalmic Surgery, Lasers and Imaging Retina, (1990); 21(11): 794-798.
- Wagle NS, Freedman SF, Buckley EG, Davis JS, Biglan AW. Long-term outcome of cyclocryotherapy for refractory pediatric glaucoma. Ophthalmology, (1998); 105(10): 1921-1927.
- Al-Haddad CE, Freedman SF. Endoscopic laser cyclophotocoagulation in pediatric glaucoma with corneal opacities. Journal of American Association for Pediatric Ophthalmology and Strabismus, (2007); 11(1): 23-28.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0
![]()

