

Review Article
Recent Advances in the Function of Vitamins in the Preventative Measures and Cure of Alzheimer’s Disease: A Literature Review
Bazza Sohail*, Abdul Wasay Nafe, Robina Malik
Adv. life sci., vol. 9, no. 3, pp. 239-269, October 2022
*– Corresponding Author: Bazza Sohail (Email: bazzasohail@yahoo.com)
Authors' Affiliations
Abstract![]()
Introduction
Methods
Discussion
Conclusion
References
Abstract
Main objective of this review is critically evaluate the use of vitamins in the safety measure as well as treatments of Alzheimer’s disease (AD) using recent findings of research on both humans and animals. The authors combed through the fundamental literature on association of vitamins (fat-soluble vitamins, water-soluble vitamins and some quasi vitamins) with prevention/treatment of AD to create this narrative review. An online search was conducted in Science Direct, Google Scholar, EBSCO & PubMed, a manual search via catalogues and scientific journals for review the literature regarding the relationship between vitamins and AD and to comprehend the potential mechanisms that may clarify such a relationship. After evaluating the titles and abstracts of the preliminary 500 articles, 350 were chosen. After reading the full texts and accomplishing a supplemental search on "Related citations" and the references of the chosen articles, a total of 282 references were found. The two most frequently mentioned ways that vitamins may affect cognitive function in AD are (1) by influences on a particular plasma amino acid called homocysteine (Hcy) for B vitamins (B6, B9 & B12), and (2) through antioxidant effects, decreasing the free radicals’ contents which can impairment human cells, for vitamins B2, C, D, E, as well as ß-carotene. These theories concerning potential mechanisms are mostly based on preclinical research and findings of links between dementia or cognitive decline and elevated Hcy levels or elevated markers of oxidative stress. The results from current studies show an effectiveness and safety of vitamin supplementation to avert mild cognitive impairment (MCI) as well as the initial phases of AD will likely rely on firstly which metabolic pathways are affected, secondly which vitamins are lacking and could correct the pertinent metabolic flaw, and thirdly, the modulating effect of nutrient-nutrient and nutrient-genotype interaction. Further emphasis on an accurate dietary strategy is needed to recognize the complete prospective of vitamin therapy in avoiding dementia and cognitive function as well as to avoid causing devastation.
Keywords: Asia, body mass index; Obesity; Physical Activity; Prevalence; World Health Organization
Introduction![]()
Alzheimer's disease is a cute, persistent and ongoing neurodegenerative disorder linked to memory loss and cognition decline leading to deceased [1]. The deposition of protein in the brain as amyloid beta-peptide plaques and neurofibrillary tangles, which produce hyperphosphorylated tau (τ)-protein and cause the death and loss of neuronal cells, are the primary pathological hallmarks of this disease [2]. Moreover, brain of such disease patients’ displays steady confirmation of oxidative stress interfere injury and prevalent inflammation [3]. Alois Alzheimer, a German psychiatrist and neuropathologist, originally identified this illness in 1906. The eponymous syndrome is now widely acknowledged as a major cause of morbidity and deaths in the ageing population, placing a significant strain on healthcare systems [4]. According to the World Alzheimer Report (2015), there are 46.8 million AD and other dementia sufferers worldwide, and that number is expected to rise to 74.7 million by 2030 and 131.5 million by 2050 [5]. This calculated rate is three to four times higher in emerging nations than it is in wealthy nations. The cost is enormous; it is reasonably projected to be above $200 billion every year. [6]. Consequently, there is a critical require to create therapies to contest AD. Numerous epidemiological studies have revealed that women are more likely than men to get AD beyond the age of 85 because women live longer than men [7]. Three clinical stages of AD have been identified like pre-symptomatic stage, pre-dementia stage and clinically characterized dementia stage [8]. The major sign of AD is the loss of short term memory and as the disease progress which cause to reduce in the cognitive behavior of the affected individual [9]. Though majority of the studies primarily interested on the late onset AD but current research studies have also initiated focusing on the early onset AD [10].
Numerous investigations have been carried out regarding its risk factors and potential causes. But there is still a lot to learn about the factors that lead to the development of AD, five hypotheses have been made comprising isoprenoid change, calcium hypothesis, amyloid hypothesis, τ-hypothesis, and cholinergic hypothesis [11]. In addition, the cholinergic theory garnered the most attention among the five hypotheses, and acetylcholinesterase is the cholinergic hypothesis’ most potential target for the therapy of AD. Currently, no cure is available for AD, which is often referred to as the ‘Plague of Twenty-First Century’ and therapy approaches concentrate on symptom reduction and behavioral change management [12].
The dietary strategy to stop, delay, or prevent the development of AD is measured to be a potential approach and has consequently been extensively researched [13]. Since nutrients are crucial for maintaining normal systemic processes, extensive study is being done to determine how they relate to the neurodegeneration that occurs in AD patients’ brains. In actuality, a number of nutrients contribute to biochemical processes and serve as an energy source for activities [14]. Because of this, dietary changes have an impact on how the central nervous system (CNS), amyloid precursor protein (APP) metabolism, and neurodegeneration are affected [14]. The factors associated with nutritional patterns believed deem important are antioxidants, calorie restrictions, hyperhomocysteinemia, and the association between apolipoprotein E (APOE) 34 genotype as well as cholesterol.
Since no treatment can halt, reverse, or even delay the neurodegenerative process, currently available medications can only temporarily improve cognitive signs. According to studies, the best nutrition and exercise practices may be the major treatment plan for AD. There have been studies looking at vitamin supplementation as an adjuvant intervention, and some of them have shown promise [15]. Since vitamins have a wide variety of functions in the CNS, they may have a variety of effects on the pathophysiological mechanisms underlying dementias. Vitamin A may help to keep beta-amyloid fibrils stable [16]. Vitamin D may help older persons’ cognition and is a precursor to hormones needed for the metabolism of calcium and phosphorus [17]. Vitamin E is an antioxidant that offers safety against harm of free radical [18]. Vitamin B-complex in particular vitamins folic acid (B9) and cobalamin (B12) play a key function in the CNS's energy production and metabolism. Additionally, vitamin B complex has been linked to the synthesis of nucleic acids and the development & maintenance of myelin, both of which are necessary for the health of neurons [19,20].
Vitamins, among other nutrients, may play a broad function in the therapy and management of AD, a systematic analysis found that the plasma levels of vitamin B9, and vitamins B12, C, and E, were considerably lower in AD populations [21]. Exogenous antioxidants that break hazardous chain reactions, such as vitamin E (tocopherol), vitamin C, and retinoic acid (RA), reduce free radical-mediated harm in neuronal cells and hence help to prevent dementia development in mammalian cells [22]. Additionally, vitamin D has antioxidant capabilities that reduce damage caused by free radicals to brain cells, preventing dementia and cognitive decline [23]. In addition, nicotinamide, the amide form of the vitamin niacin (B3), is essential for cellular function and the metabolism of energy because it is the precursor to the coenzyme nicotinamide adenine dinucleotide (NAD+). In an AD animal model, nicotinamide therapy has been shown to avoid cognitive losses while enhancing short-term spatial memory, demonstrating its potential as an AD therapeutic [24].
The electron transport chain (B2, B3, and B5), 1-carbon metabolism (B2 and pyridoxine or B6), and methylmalonyl-CoA mutase (B12) all require a number of vitamins as cofactors, as is clear from basic biochemistry. These include the citric acid cycle, which includes thiamine (B1), riboflavin (B2), pantothenic acid (B5), and biotin (B7) [25]. It is necessary for the vitamin B12 to get energy in the form of succinyl-CoA from methylmalonyl-CoA, which is predominantly generated from propionyl-CoA, a compound created by the catabolism and digestion of isoleucine, valine, threonine, methionine, thymine, cholesterol, or odd-chain FAs. This suggests that restoring deficiencies in the vitamins B1, B2, B3, B5, B6, B7, B9, and B12 may increase mitochondrial metabolism in the aged brain, perhaps increasing the efficiency of adenosine triphosphate (ATP) synthesis.
The vitamins that have been linked to AD in human epidemiological and interventional investigations are the main topic of this review. In order to prevent AD with vitamin therapy, it identifies significant knowledge gaps and offers new research pathways based on precision nutrition.
Methods![]()
Literature survey and selection criteria
This review included articles from different databases, manual search of journals and bibliographies, as well as use of PubMed, Science Direct, EBSCO, Google Scholar, and other sources. The review was performed in Pakistan, at Riphah International University, Islamabad.
Investigate Requisites
Three key elements emerged from the research question: Alzheimer's disease, vitamins, and the way the later one responded to the first. In order to create a search technique that is used in all database searches, synonyms for the terms Alzheimer's disease, "dementia," "cognitive," and vitamins were all taken into consideration. Title, abstract, and full text readings have all been done before reading any studies.
Criteria for Inclusion and Exclusion
Initially, only articles from peer-reviewed journals were included; proceeding articles, reviews, editorial content, meeting abstracts, letters, retracted publications, and book chapters were all removed. Both observational and interventional study types were included in terms of study designs. The articles were exclusively available in English, which allowed for the most accurate data extraction. Additionally, a majority of articles within the last 10 years were included in order for the research to be practical. Lastly, the review included the most studies involving human subjects possible in order to provide a higher level of evidence.
Figure 1 shows the scoping review flow diagram. After examining the titles and abstracts of the initial 500 publications, 400 were chosen. After reading the full texts and running a supplemental search on "Related citations" and the references of the chosen articles, a total of 282 references were found..
Humans generally need enough levels of nine water-soluble vitamins, including vitamin C and the eight B vitamins [thiamine (B1), riboflavin (B2), niacin (B3), pantothenic acid (B5), pyridoxine (B6), biotin (B7), folate (B9), and cobalamin(B12)], as well as four fat-soluble vitamins (A, D, E, and K). Primarily the former bind to cellular nuclear receptors and influence the expression of particular genes [26]. The latter primarily function as an enzyme's cofactor and influences enzymatic activity.
Fat-Soluble Vitamins
Vitamin A and beta-Carotene
The most versatile natural retinoid, vitamin A (retinol), affects a wide range of biological activities, including normal brain function, cell differentiation, cell growth, embryonic development, and apoptosis [27]. Pro-vitamin A carotenoids, which are found in a variety of colorful fruits and vegetables (such as kiwi, carrots, tomatoes and green leafy vegetables), as well as some animal sources, are typically used to make retinol (liver, egg yolks, or dairy products). Carotenoid hydrocarbons are the group to which beta-carotene belongs. It performs as an antioxidant because of its nature of scavenger and quencher, and its collaborative impact with other antioxidants [28].
Both vitamin A and beta-carotene may be essential molecules for the halt and treatment of AD, because of their capability to reduce the creation of both A-beta oligomers and fibrils [29]. Vitamin A and beta-carotene have been found to have anti-oligomerization effects on A-beta in vitro [30]. Vitamin A and beta-carotene levels were shown to be low in the serum and plasma of AD patients, and a higher plasma level of beta-carotene was associated with improved memory function [31]. Carotenoids’ anti-oxidant abilities have mostly been documented [32], while other mechanisms, including anti-inflammatory functions, modification of the structural and functional characteristics of synaptic membranes, control of cell growth, improvement of gap junctional communication, conversion of beta-carotene to vitamin A, and enabling the modulation of gene expression, may also partially contribute to the beneficial biological effects of carotenoids on the brain [33]. When it comes to dementia, relationships between plasma beta-carotene concentrations and cerebrospinal fluid (CSF) levels of A-beta-40 and -42 and total τ, which are neurochemical indicators of AD, have been found among 40 AD patients, suggesting a potential beta-carotene anti-amyloid action [32].
Retinol, the most bioavailable form of vitamin A, is transformed by the body into retinal and retinoic acid (RA). The transport of retinoid from the gut to target organs, including the brain, has been observed to be altered in AD. Vitamin A and all retinoids—natural or synthetic—have a chemical connection. Although the dissemination of RA inside a mature human CNS is unknown, sampling live human brains is impossible, and RA degrades quickly in autopsied brains, there is indirect proof that suggests decreased RA contents in AD brains [34]. Retinaldehyde Dehydrogenase (RALDH), the enzymatic synthesis of RA, has been discovered to be higher in AD brains, which supports the speculation that AD patients have reduced concentrations of RA. Higher concentrations of RALDH have been exhibited in cases where neuronal cell lines have been deprived of retinoid. But, in healthy patients, the injection of retinol would have brought these elevated levels back to normal through feedback processes [35].
One of the most crucial objectives in the therapy of AD is the prevention of neuroinflammatory reactions. Significant retinoid inhibition of IL-6 production suggests that retinoids may be an effective treatment for AD [36]. By preventing the nuclear translocation of nuclear factor-kappa B, retinoids have been found to reduce lipopolysaccharide-induced or A-beta-induced tumour necrosis factor-a production and to hamper appearance of inducible nitric oxide synthase in activated microglia [37]. All-trans-retinoic acid (ATRA) is used to make synthetic retinoids, which are intended to have strong therapeutic efficacy and no negative side effects in clinical setting. 13-cis-retinoic acid was the first synthetic retinoid to be licenced for clinical use orally in the US (isotretinoin). More recent studies also show that ATRA inhibits lipopolysaccharide-induced neuroinflammation, amyloid genesis, and memory deficits in old rats [38] and promotes proliferation of neural stem cells along with suppression of activation of microglia leading to adult neurogenesis in the hippocampus in a mouse model of AD ATRA restrains lipopolysaccharide-induced neuroinflammation, amyloid genesis, and memory shortage in old rats, according to more recent research [38], and it encourages neural stem cell proliferation and suppresses microglia activation, which results in adult neurogenesis in the hippocampus (the part of the brain that stores mental maps) in a mouse model of AD [39]. According to a fairly recent study, ATRA reduced neuroinflammation in the rat brain via altering the expression of nuclear factor-kappa B and Sirtuin 1, two proteins that belong to the Sirtuin family of class III histone deacetylases [40].
Retinoids may have therapeutic benefits for neuroprotection, which would stop the pathogenesis of AD. Vitamin A and other retinoids can directly block the formation of A-beta plaques in vivo [41]. It has been proposed that RA can substantially potentiate neurotransmitter functions of acetylcholine in cholinergic neurons in the brain. Acetylcholine’s neurotransmitter actions in cholinergic neurons in the brain have been suggested to be significantly enhanced by RA [42]. Cognitive and memory problems are a result of cholinergic neuron degeneration. The use of RA and its derivatives in the treatment of AD may be justified by their cholinotrophic characteristics [42]. The co-activation of RA receptors and (with the agonist Am80 or Tamibarotene) and retinoid X receptors was necessary for an effective therapy of AD in a mouse model (with the pan agonist HX630) [43]. In this study, it was found that giving APP23 mice Am80 (0.5 mg/kg) and HX630 (5.0 mg/kg) together for 17 days significantly restored their memory deficiencies, whereas giving either drug alone had no therapeutic impact. These researchers did not, however, note any possible negative consequences of combining RA receptor and retinoid X receptor agonist therapy.
To control ongoing stem cell turnover, cell plasticity, and tissue regeneration processes, natural and synthetic retinoids and their receptor agonists are being studied. Retinoid signalling dysfunction encourages AD pathogenesis. Retinoids are intriguing therapeutic prospects since they are tiny molecules that can easily reach the tissues. Their use at lower doses or in combination with other neuroprotective medications could reduce unintended adverse effects in tissues other than the ones intended. Thus, retinoids constitute a novel therapeutic approach for the treatment of AD, as they can inhibit a number of the illness's clinical symptoms, such as plaque development, neuroinflammatory responses, and brain neurodegeneration [41].
Vitamin D
In the 20th century, vitamin D was first thought of as a vitamin; today, it is understood to be a steroidal hormone. Critical brain activities and the development of the brain (effects on cellular proliferation, differentiation, calcium signaling, neurotrophism, and neuroprotection) are both impacted by it [44]. It comes in vitamin D2 and D3 isoforms. The terminal calcitriol-activating enzyme, 1, a-hydroxylase, and vitamin D receptors are found throughout the adult and foetal brain. Przybelski discovered a significant association between serum 25(OH)D levels and cognitive scores [45]. According to Sommer and coworkers [46], the epidermis produces around 90% of the body’s vitamin D from 7-dehydrocholesterol in response to sunshine (solar ultraviolet B radiation; 290-315 nm). Diet, fortified foods, and supplements are some additional sources. Biologically inert at first, vitamin D requires two independent hydroxylation reactions to transform into its active form. First to 25(OH)D in the liver, then to 1,25-dihydroxyvitamin D in the kidney, which is the active hormonal form [47]. A 25(OH)D level of less than 50 nmol/L is typically considered to be deficient in vitamin D, with severe deficiency being defined as fewer than 25 nmol/L and insufficiency as between 50 and 75 nmol/L [48]. According to these standards, around 1 billion people globally have insufficient or inadequate vitamin D levels [48]. For a number of reasons, elderly people are more vulnerable to vitamin D shortage. For instance, as people age, their skin becomes thinner, which lowers the skin's quantities of the precursor to vitamin D3 called 7-dehydrocholesterol [48]. Other causes include inadequate sun exposure, a reduction in vitamin D intake from food, poor intestinal absorption, and poor hydroxylation in the liver and kidney [47].
Low serum vitamin D levels may be linked to AD, various kinds of dementia, and cognitive impairment, according to recent systematic reviews and meta-analyses from cross-sectional studies [46,49,50]. In a meta-analysis of eight studies with a combined total of 2,749 participants, Balion and colleagues [49] found that participants with serum 25(OH)D deficiency (<50 nmol/L) performed on average 1.2 points worse on the Mini-Mental State Examination, a test of general cognitive function, than participants with serum 25(OH)D sufficiency (>50 nmol/L). In seven case-control studies, Annweiler and colleagues [51] discovered that the serum 25(OH) D levels of 357 AD patients were 1.4 standard deviations lower than those of 648 controls. A further meta-analysis study [52] found that patients with low levels of vitamin D (25(OH)D level 50 nmol/L) had a 21% higher chance of getting AD than those with levels of 25(OH)D level >50 nmol/L). A considerably higher incidence of dementia was discovered in people with vitamin D deficiency by same study. Similar to this, Sommer and others [46] stated that a meta-analysis of five studies revealed an increased risk for those with severe vitamin D shortage (<25 nmol/L or 7-28 nmol/L) compared to individuals with adequate vitamin D supply (≥50 nmol/L or 54-159 nmol/L). However, other systematic reviews [53] were unable to identify a relationship between 25(OH)D levels and cognitive function as determined by the Mini-Mental State Examination. Systematic review results may differ depending on search methods, inclusion criteria, statistical analysis methods, and confounding factor adjustments. Because they frequently control for confounding factors and provide information on the time sequence of cause and effect, longitudinal studies under the category of observational studies may be more reliable than other observational study designs.
According to six prospective studies, low vitamin D levels are strongly linked to an increased risk of cognitive decline [54,55,56,57,58] or diminished cognitive performance [59]. On the other hand, among vitamin D quartiles tested at baseline in 1,604 males aged 65 and older, there was a non-significant monotonic rise in the odds of overall cognitive deterioration on the Modified Mini-Mental State Examination over a mean follow-up time of 4.6 years [55]. Likewise, there was no correlation between vitamin D tertiles and incident global cognitive impairment on the Standardized Mini-Mental State Examination across a 3-year period in 299 adults aged 85 years and older [60]. The results of a systematic review and meta-analysis on the relationship between vitamin D levels and particular cognitive functions, however, point to a strong correlation between low vitamin D levels and a number of executive dysfunctions, including sluggish processing speed, mental shifting, and information updating [50].
Four studies have examined the impact of vitamin D supplementation on elderly people's cognitive outcomes; three of the studies [61,62,63] were interventional studies, and the fourth [64] used a post-hoc approach. Overall, three investigations indicated that vitamin D administration did not lower the risk of dementia or MCI relative to controls [64] or enhance cognitive outcomes[61,62,64]. Contrarily, a study indicated that participants who got vitamin D3 supplementation (800 IU per day or 100,000 IU per month) over a 16-month follow-up period had enhanced executive functioning and global cognition as compared to controls [63]. The results of the interventional studies are challenging to interpret, however, due to methodological flaws like small sample sizes [61,62,63], brief follow-up periods [61,62], a lack of participant randomization [61,63], low baseline vitamin D levels and varying vitamin D supplementation doses. The post-hoc analysis also contains a number of significant flaws [64]. The trial was not suited to examine the benefit of vitamin D supplementation on dementia risk because it used a post-hoc methodology.
The relationship between vitamin D and dementia-related outcomes in senior persons has been examined in five trials, with four assessing serum 25(OH) D concentrations [65,66,67,68] and one assessing vitamin D food consumption [63]. Such increased vitamin D food consumption was linked to a lower risk of AD but not non-AD dementias above 7 year follow-up duration in the latter trial, which involved 498 females with a mean age of 79.8 years [63]. Higher 25(OH)D levels were linked to a lower risk of dementia-related outcomes, according to three of the four studies that assessed 25(OH)D contents [65,66,67]. Over the course of seven years, severe vitamin D deficiency (<25 nmol/L) in 40 high-functioning senior women aged 75 years or older was associated with a greater risk of non-AD dementias but not AD [65]. On the other hand, across a median follow-up of 21 years, severe vitamin D deficiency (<25 nmol/L) was associated with AD but not vascular dementia in 10,186 subjects [66]. The first study had a very small sample size, which probably contributed to its lack of statistical power [65], while the second study relied on diagnoses of dementia from non-standardized medical records, which might have led to significant misclassification [66]. Two recent high-quality studies [59, 69] that looked at four recent high-quality studies [70, 71, 72, 73] revealed no correlation between 25(OH)D concentrations and dementia or AD.
In the first investigation into a potential genetic link between AD and the vitamin D receptor (VDR), researchers found that a polymorphism in the VDR area more than doubles the risk of AD [74]. They also stated that some of the changes in the vitamin D-VDR pathway may be caused by single nucleotide polymorphisms in the VDR gene. Kuningas and coworkers [75] reported that the genotyping of 563 participants (over 85 years old) for five distinct variants in the VDR gene revealed a connection between gene variation and age-related alterations in cognitive performance. In contrast to carriers of the ApaI variation, carriers of the BamI and TaqI polymorphisms demonstrated lower cognitive performance. Another study showed ApaI and TaqI gene polymorphisms in 260 cognitively tested older controls and 255 AD patients and found a correlation between the existence of each of these haplotypes and the risk of AD [76]. Recently, 108 AD patients and 77 healthy controls from the Lower Silesian population cohort were compared to determine the frequency of the VDR polymorphisms TaqI, ApaI, FokI, and BsmI [77]. According to this study the frequency of the TaqI, Fok1, or BsmI polymorphisms was not substantially different between the two groups. But the frequency of the ApaI allele A in the control group was higher, and this later led to a 30% decreased risk of AD in studies in Polish and British populations [77]. The researchers discovered a significant variation in risk alleles for these VDR polymorphisms depending on the population they analysed, indicating a dependence on racial origin and environmental factors [77].
A single-nucleotide polymorphism identified in the transcription factor Cdx-2 binding site has been strongly linked to late-onset AD (492 AD cases versus 496 controls) [78]. The research revealed a relationship between reduced VDR promoter activity and the risk allele for CDX2 [78]. According to one study, the prevalence of the “TaubF” haplotype (alleles of the proteins TaqI, ApaI, Tru9I, BsmI, and FokI, respectively) was considerably greater in the patient group with AD than controls and can be viewed as a risk factor for the condition [74]. In a sample of US people aged 50 and older, sex-specific gene polymorphisms in the VDR and megalin genes were found to ameliorate age-related cognitive deterioration [79].
Results from different studies suggested that vitamin D may play a role in both preventing and treating AD and has a variety of roles across the CNS. Although reverse causality is still a possibility, cross-sectional and case-control studies corroborate the finding that people with cognitive impairment and dementia have lower vitamin D concentrations. In order to address this, longitudinal studies have discovered a correlation between low vitamin D contents and a higher risk of cognitive decline, all-cause dementia, and AD. Clinical research examining the impact of vitamin D addition on cognitive outcomes have yielded contradictory results; although, the applicability of these results is constrained by a number of methodological flaws. Vitamin D influences neurotransmission, calcium homeostasis, vascularization, A-beta and τ-accumulation, oxidative stress, and inflammation, all of which are disrupted in AD, through both genetic and non-genomic impacts. However, the pleiotropic effects of vitamin D are tissue-, cell-, time-, person-, pathological context-, dose-, and perhaps, gender-dependent. One hypothesis is that vitamin D is an effective therapeutic target for both preventing and treating AD.
Vitamin E
It is composed of 8 analogs: a-, beta -, gamma- & δ-tocopherol as well as a-, beta -, gamma- & δ-tocotrienol and belongs to fat-soluble vitamins. The most prevalent and accessible antioxidant form of vitamin E in human tissues is a-tocopherol. Depending on age, many countries recommend a daily dietary consumption of between 3 and 15 milligrams of vitamin E [80]. Tocopherols and tocotrienols are abundant in seeds and edible oils including those from almond, peanut, olive, palm, canola, corn, and soybean while being sparse in plant foods with low lipid levels like fruit and vegetables [81]. There is a lot of interest in this vitamin as a result of the development of the free radical theory to explain the pathophysiology of brain ageing and neurodegenerative illnesses, such as AD. This fat-soluble vitamin possesses anti-inflammatory, hypocholesterolemic, and neuroprotective characteristics that contribute to its significance for brain health [82]. Additionally, vitamin E, particularly the a-tocopherol form, is regarded as one of the most significant antioxidants in the brain. This is because the transporter a-tocopherol transfer protein (a-TTP), whose roles include regulating and distributing vitamin E concentrations in various tissues, is found in high concentrations in the brain[83]. The cerebellum of the encephalon exhibits the highest levels of a-TTP expression, particularly in astrocytes, which give nearby neurons vitamin E [84]. Prominently, people with neurodegenerative illnesses have higher levels of a-TTP expression in their brains [85]. As a result, it is possible to draw the conclusion that vitamin E's antioxidant activity plays a significant role in neuroprotection. Owing to its strong antioxidant capabilities and the biological plausibility of its possible function in preventing the degenerative processes underlying AD, vitamin E has received a great deal of research attention. Though, there is debate regarding whether or not it is a useful clinical intervention.
Beyond its function as an antioxidant, vitamin E helps older people's immune systems. A dysregulation of the immune system that results in numerous inflammatory responses is developed during ageing and in AD. Numerous studies show that vitamin E, taken as a dietary supplement by aged people, has anti-inflammatory properties. These investigations demonstrate, among other positive outcomes, in vitro T-cell proliferation, IL-2 generation, and E2 prostaglandin inhibition [86,87]. A study showed an enhancement in IL-2 receptor levels in T-lymphocyte population when with 233 mg of vitamin E each day for 28 days [88]. Another study also revealed that aged persons who received a-tocopherol at a similar quantity (233 mg) for three months noted improvements in their lymphocytes’ ability to adhere, their body’s ability to produce IL-2, their natural killer activity, and the proliferation of their lymphocytes [89]. The immune system is influenced by vitamin E through the suppression of prostaglandins E2 and D2, but not by the actions of cyclo-oxygenase and 5-lipoxygenase [90].
Many trials revealed lower plasma level of vitamins E in AD patients. Some scientists performed a meta-analysis and evaluated eighty studies on micronutrients as well as AD [91]. They found that patients with AD had reduced plasma levels of, among other things, vitamin E. Additionally, they found no correlation between the patients’ vitamin E levels and their malnutrition condition, and they hypothesized that a compromised micronutrient status may occur prior to malnutrition. Vitamin E concentrations in CSF and brains are considerably lower in AD patients, according to a recent meta-analysis of 116 articles [92]. A more recent [93] meta-analysis involving 17 trials, 904 AD patients, and 1153 healthier older controls revealed that AD patients had lower serum vitamin E concentrations than healthy older controls. A new Mendelian randomized study examined the association between circulating vitamin E and AD, but some investigations found no difference in vitamin E levels [94]. The researchers examined information from a large-scale GWAS (genome-wide association study) on vitamin E that included 7781 people of European ancestry in total, as well as a GWAS that included 17,007 AD patients and 37,154 controls. This study exhibited no substantial correlation between AD and concentration of vitamin E.
Regarding epidemiological research, a link between vitamin E consumption and a lower chance of developing AD was also revealed. A study found a clearly decreased incidence of AD in persons taking vitamin E and multivitamin complexes that also contained vitamin C [95]. It’s interesting that researchers couldn’t find any proof that taking these chemicals alone had any protective effects. Additionally, 365 AD patients’ data that were examined revealed a slight decline in the long-term risk of AD [96]. Notably, the favorable outcomes were only seen in people who consumed more foods high in vitamin E [96]. Participants who consumed an average amount of vitamin E did not, however, have a lower risk of dementia [96]. The usage of vitamin E supplements was linked to a lower incidence of cognitive decline, according to data from the Canadian study of health and ageing (1991-2002), a cohort study of dementia involving 560 AD patients [97]. Three other investigations, however, did not find any correlation between vitamin E intake and the chance of developing AD [98.99,100]. The first study, which involved 2969 participants and was followed up on every two years for 5.5 years, came to the conclusion that taking more vitamin E and C either separately or together did not lower the incidence of AD or general dementia [98]. The second study, which included 3385 men from the Honolulu-Asia ageing project, exposed that vitamin E and C supplementation may enhance cognitive performance in old age, but that there was no protective impact for any specific form of dementia, including Alzheimer’s [99]. Finally another research with 980 older participants in the Washington Heights-in-Wood Columbia Aging Project revealed no link between vitamin E intake, whether it is dietary, supplementary, or total, and a reduced incidence of AD [100].
In a three-year, double-blind trial, 769 participants were chosen and given either a placebo or 2000 IU/d of vitamin E. It was found that supplementing with vitamin E had no positive effects [101]. Another study revealed no differences in cognitive function between vitamin E-treated females and those who received a placebo [102]. It is intriguing to note that the vitamin E group, which comprised women whose daily vitamin E intake was below 6.1 mg, showed less cognitive impairment relative to the placebo group than did the women in the high vitamin E intake group [102]. In a different study [103], participants received 800 IU/d for six months, and the vitamin E supplemented group was split into two distinct subgroups. One group, referred to as “respondents” to vitamin E, experienced decreased oxidative stress measures following the treatment while maintaining their cognitive test results [103]. They did ascertain a second group of individuals, referred to as “non-respondents,” in which vitamin E had no effect on reducing oxidative stress and cognition had dropped to levels even lower than those of patients receiving a placebo [103]. It was determined that patients’ cognitive condition is maintained when vitamin E reduces oxidative stress. However, oxidative stress is harmful to cognition when vitamin E does not stop it. On the other hand, a study that involved 613 patients with mild to moderate AD and was double-blind, placebo-controlled, parallel-group, randomised, and used the same dose (2000 IU/d of a-tocopherol or placebo) displayed that a-tocopherol supplementation decreased functional decline in patients with mild to moderate AD [104]. The groups receiving memantine alone or memantine with a-tocopherol showed no discernible differences [104]. These results demonstrated that a-tocopherol is beneficial for mild to moderate AD by delaying functional decline and reducing caregiver stress. A recent study indicated that supplementing with 400 IU/day of vitamin E did not prevent dementia in selected 7540 asymptomatic older males [105]. The research also demonstrated that neither selenium nor vitamin E, or even a combination of the two, could prevent dementia [105].
Moreover, efforts to develop a vitamin E-based AD therapy have ongoing. Numerous research are still being conducted to determine how well certain vitamin E forms work alone or when combined with another substance that may slow the progression of dementia. In a transgenic mouse model of AD, Ibrahima and coworkers [106] examined the therapeutic effects of the tocotrienol-rich fraction, a mixture of several vitamin E equivalents from palm oil. The findings demonstrated that supplementing restored cognition function in transgenic mice and decreased A-beta deposition even if the amounts of A-beta oligomers remained constant. Last but not least, a further investigation using an animal model of AD examined the effects of supplementing with fish oil and vitamin E [107]. The findings demonstrated that the transgenic mice’s cognitive function could only be restored by a diet deficient in vitamin E and a fish oil supplement [107]. Higher vitamin E dosages had no positive effects, indicating that excessive vitamin E acts as an oxidant and promotes oxidation [107].
The varying and contradictory outcomes that numerous researches have shown may be influenced and explained by a number of factors. First, it's crucial to assess the dose of vitamin E as a variable. High doses of vitamin E were utilized to achieve the positive outcomes. Subsequently in some studies, the dose of vitamin E utilized was insufficient to produce a therapeutic outcome. Although further research is required to identify the ideal amount, it is crucial to note that greater doses do not appear to be linked to higher mortality rates or adverse consequences. Secondly, the type of vitamin E utilized is another crucial consideration. While most vitamin E supplements contained alpha-tocopherol, it appears that other dietary tocopherols and tocotrienol, as well as their combination, may be more effective in achieving a greater neuroprotection. Timing the start of the study is another issue that needs to be addressed. Given that no study has shown the ability to reverse the illness process, it is conceivable to postulate that an early administration may be more beneficial in preventing cognitive losses. According to this viewpoint, the severity of AD is a crucial consideration that may affect the outcome of the therapy. As well, it appears that ongoing therapy is required to achieve a noticeable improvement. Also, it must be remembered that not all studies used the same criteria to assess whether the vitamin E treatment was effective, making it impossible to compare the findings of other studies. In fact, whereas some studies assessed the ability to carry out daily tasks, others believed the development of MCI into AD into account. There is impossible to rule out confounding factors that could affect the results, like the usage of other medications or the existence of coexisting conditions.
It may be concluded that despite a compelling case for vitamin E's use as a successful AD intervention, the clinical studies that have been conducted to date have produced unreliable results regarding vitamin E's impact on the risk of acquiring AD. Therefore, it is yet unknown whether vitamin E concentrations are genetically linked to an increased risk of AD or whether taking supplements of this substance could help slow the course of dementia. In this review, it was shown that there are more studies demonstrating a drop in plasma vitamin E levels in AD patients than studies demonstrating the opposite. Vitamin E as a medication does, but occasionally improves cognition and other times it does not. The replacement of lost neuronal networks, the very different dietary status of the patients at baseline, the varying lengths of time for brain compensation in each individual, and the antioxidant effect of vitamin E in each individual, among other factors, could all contribute to treatment failure. Though, more research is needed to provide definitive answers about vitamin E's effectiveness in AD. Trials comparing various vitamin E dosages, forms, and combinations may be beneficial in this situation.
Vitamin K
The primary dietary source of vitamin K is phylloquinone, and vitamin K-2, known as menaquinones, contains a number of vitamins with bacterial basis [108]. In human and rat brains, menaquinone-4 (MK-4) is the vitamin with the highest representation [109]. In-vitro research has shown that MK-4 shields against oxidative damage and the activation of the inflammatory cascade [110,111]. Moreover, it has been discovered that MK-4 depletion in murine models is associated with lower cognitive abilities [112]. As a cofactor of G-glutamyl carboxylase, vitamin K is known for its function in blood coagulation and the activation of factors that depend on it, including factor II, VII, IX, X, protein C, and protein S. Vitamin K may play a function in brain physiology by contributing to the metabolism of sphingolipids and by activating the vitamin K-dependent growth arrest-specific 6 protein (Gas6), which may prevent neuronal death brought on by amyloid or oxidative stress [108]. This vitamin K-dependent gene is thought to contribute to oligodendrocyte survival and, as a result, increase myelination in the CNS. Sphingolipid synthesis, which appears to be crucial for remyelination, is likewise greatly aided by Gas6.
There is considerable disagreement as to whether vitamin K insufficiency contributes to cognitive deterioration in AD patients. Human studies [113-118] were found in the literature search; all, with the exception of one [115], confirmed a connection between vitamin K and cognitive performance in older persons. In a population of 65 years and older, five investigations found a connection between declining cognitive and behavioral abilities and low vitamin K food intake or serum level. In particular, researchers reported the findings of a cross-sectional study done on 320 older people from the Nukage study cohort, aged 70 to 85, who were cognitively healthy [114]. According to studies on food intake in older persons, 1) those with major subjective memory complaints consumed less vitamin K on average than those with normal memory (298.0 mg/d against 393.8 mg/d) [117], 2) on a person-day basis, the mean vitamin K intake in individuals with early-stage AD was 63 g/d, as opposed to 139 g/d in 31 healthy control persons [114], and 3) among older adults who took part in the CLIP trial, higher dietary phylloquinone consumption was positively linked with better cognitive function [116]. The frontotemporal behavioral rating scale (FBRS), a measure of physical neglect, and dietary vitamin K levels were found to be negatively correlated with each other in the scientist’s observational cross-sectional study, which examined the relationships between neuro-cognition and lipophilic vitamins among all patients consecutively hospitalized or seen in consultation in the geriatric acute care unit of the University Hospital of Angers, France, from February to April 2014; the FBRS is a consistent, reproducible marker of the presence of signs of behavioral disturbance across four domains (i.e. self-control disorder, physical neglect, mood disorders and loss of general) [116].
One study that measured plasma levels of vitamins K-1 (phylloquinone) and K-2 (menaquinone) found that patients with mild and severe AD had considerably lower plasma vitamin K-1 concentrations than healthy controls, but that there was no difference in plasma vitamin K-2 between cases and controls [113]. We are not aware of any intervention studies that have looked at how vitamin K deficiency or supplementation affects cognitive performance or stops brain atrophy. Other research investigated the relationship between cognitive impairment and the use of oral anticoagulants known as vitamin K antagonists (VKAs), such as warfarin, acenocoumarol, and fluindion [119,120]. In fact, these VKAs carry out their purpose by inhibiting the activity of vitamin K oxidoreductase, which stops vitamin K from being recycled after being gamma-carboxylated. It has not yet been shown if VKAs affect Gas-6 and protein S gamma-carboxylation in the human brain. Studies using murine models have demonstrated how VKAs predict a drop in MK-4 brain concentrations, the most recognizable vitamin in rats’ brains [121]. Since anomalies of the CNS were determined in babies who had been exposed to warfarin or other coumarin derivatives, it has been clear that VKAs may have a negative effect on the brain. [120]. Absolute mechanisms, though, are still unknown. How the usage of VKAs may affect brain metabolism is a growing concern. The scant literature suggests, to a limited extent, a potential link between the usage of VKAs and both brain focal atrophies and cognitive decline. During a big cohort trial, there was no correlation found between the use of VKAs and global cognitive performance over a 10-year follow-up period, however there was a substantial decline in verbal fluency and visual memory among patients treated with VKAs compared to those not receiving blood-thinning medication [121]. Researchers used 3 or 1.5 Tesla magnetic resonance imaging (MRI) to measure the brain volumes of 54 individuals (18 receiving VKAs treatment and 36 matched controls) and found a significant inverse relationship between the length of drug exposure and the volume of grey and white matter in the right precuneus, left middle frontal gyrus, and right inferior operculum in the right front [122]. Clinical investigations [123] have revealed that VKA use has no effect on vitamin K plasma levels, and that there are still unexplored molecular patterns that could cause cognitive impairment in VKA-using human. When suggesting that VKAs are responsible for cognitive impairment, it is important to keep in mind that patients may be receiving therapy for conditions like atrial fibrillation, which may have a minimal impact on mental decline [124].
The lack of published articles points to the require for more thorough investigation from the scientific community, using large samples in randomized trials to support the concept that low vitamin K levels may be associated with cognitive impairment. To produce more comparable and trustworthy data, a systematic methodology for measuring vitamin K intake from diet and blood concentrations must be implemented. A large prospective study is feasible and could be necessary to clarify the impact of these medications on vitamin K blood levels and, subsequently, on cognitive impairment because of the vast number of people who have received VKA treatment.
Overall findings indicated a robust association between fat-soluble vitamins and AD. According to reports, a lack of vitamins A, D, E, and K is closely related to AD. On the other hand, higher levels of these vitamins in the serum or plasma have been associated with improved cognitive abilities. Hypovitaminosis of vitamins D and E appear to be a risk factor for the development of AD, and there are some signs that supplementing with these vitamins may be helpful in the uncertain progression of AD. The hypothesis that the supplementation of these compounds might be advantageous is strengthened by the observation that deficiencies of vitamins A, D, E, and K lead to elevated cerebral A-beta levels and/or impaired cognitive abilities in animal models, while the supplementation of vitamins A, D, and E reduces A-beta plaque load. Vitamin E supplementation, in particular, may have certain negative effects. For instance, several studies have found that AD patients have a risk for A-beta-increasing and even more rapid cognitive loss.
Briefly, more research is needed to assess the potential of these substances in the prevention and treatment of AD, particularly major trials looking at the effectiveness of various vitamins in AD patients and their impact on the molecular mechanisms of the illness.
Water-Soluble Vitamins
Vitamins B complex/multi Vitamins
With the exception of vitamin B3, which may also be produced from tryptophan, almost all members of the B family of vitamins are necessary forms that depend on diet provide. Studying the correlation between brain atrophy, metabolic activities (such as glucose metabolism), and amyloid buildup, which can be assessed using brain MRI, fluoro-2-deoxy-D-glucose standardized uptake value ratio, and Pittsburgh compound B-positron emission tomography (PIB-PET), respectively, is a new straight forward method to examine the relationship between B vitamins and AD [125]. Three vitamins B1, B7, and B12 exhibit well-established relationships with the deterioration in mental status due to the three B-complex vitamins' associations with neurological diseases [126]. Patients with MCI who underwent MRI showed a substantial positive correlation between the pace of brain atrophy and Hcy level (Hcy is a biomarker of either vitamins B9 or B12 shortage, or both) [127].
Additionally, supplementing with vitamins B9 (0.8 mg/d), B12 (0.5 mg/d), and B6 (20 mg/d) for two years reduced brain atrophy in amnestic or non-amnestic MCI patients [127]. These positive benefits were predominantly observed in people with Hcy (>11 mmol/L) or plasma omega-3-fatty acids (ω-3 FAs; >390 mmol/L), and they were associated with increased vitamin B12 status and were reflected in improved cognitive ratings [128].
The vitamins B9 and B12 are recognized as two essential micronutrients in people. Fore-mentioned two vitamins are vital for DNA synthesis and repair, the metabolism of FAs and some amino acids, as well as the healthy operation of the nervous system. They are also critical cofactors in methylation processes [129]. These two vitamins are crucial cofactors in the conversion of hydroxycitric acid to methionine. One of the most typical impairments in AD patients is a shortage in B12 and B9, and in the majority of instances, elevated concentrations of Hcy are seen. Hcy metabolism plays a major role in the mechanisms underlying the relationship between vitamins B-complex and the brain. It is broken down via a remethylation cycle that is driven by vitamins B9 and B12 and provides methyl groups for a number of metabolic processes [130]. Hyperhomocysteinemia has been proposed as a key risk factor for AD because clinical proof has linked higher plasma (Hcy) levels to the development of AD [131]. Observational investigations have shown that AD risk is elevated in those with high blood levels of Hcy [130]. The outcomes of these studies, which aggravated randomized trials to assess if vitamins B complex had a positive impact on cognition, were conflicting. In the systematic review, O’Leary and colleagues identified 35 cohort studies with 14,235 people in total [132]. Only seven of the 21 high-quality studies revealed a helpful link. Studies that used the more recent and precise indicators of vitamin B12 status, such as methylmalonic acid and holotranscobalamin, found that this association was more reliable.
The most of observational studies failed to find a positive correlation. A meta-analysis of RCTs in people with or without cognitive impairment was carried out [133]. Overall, the 19 RCTs that were examined revealed that there was no effect of vitamin B supplementation on cognition. Likewise results were found in an earlier Cochrane analysis [134], which displayed that B9 treatment, whether it was given with or without vitamin B12 or B6, had no effect on persons with cognitive impairment or dementia. A subsequent Cochrane update, however, which involved 8 trials, found that there was some effect [135]. For example, in certain investigations involving both AD and without dementia patients, a greater response to cholinesterase therapy with B-complex vitamins was noted. The B9 has produced some other contradictory findings following the Coronary Intervention Trial (n=800), which claimed that vitamin B9 had a substantial impact on memory [136]. This study demonstrated that over a period of 2-3 years, those who received vitamin B9 treatment and had high levels of Hcy had higher cognitive performance than those who did not receive treatment [136]. One of the neurotropic B-complex vitamins' (specifically, vitamins B6, B12, and B9) most important functions in the CNS (i.e., the brain and spinal cord) come from their role in the metabolism of B9 and Hcy. Elevated Hcy concentrations are linked to several vitamin shortages, which are thought to have neurotoxic consequences. Excessive Hcy may enhance oxidative stress and neurodegeneration, which can raise the risk of AD, dementia, and cognitive decline [137].
The relations of vitamins B9, B12, D, beta-carotene, and ω-3 FAs with a healthier brain imaging profile are maintained by 1) findings from a meta-analysis of 43 prospective cohort studies, which showed that dietary patterns reflecting a higher consumption of vitamins B6, B9, and B12, vitamin E, unsaturated FAs, and vitamin C were related with a lower relative risk for dementia [137]. 2) the results of a meta-analysis of 106 studies on the plasma vitamin level in AD patients, which revealed considerably lower levels of the vitamins B12 (212%), B9 (221%), C (233%), A (214%), D (227%), and E (218%) than in healthy controls [138]. But it is essential to remember that investigations may not always be comparable across populations and cohorts even though dementia may indeed manifest in cohorts that are not vitamin insufficient or do not have further risk factors like obesity, hyperhomocysteinemia, or inflammation, as was, for instance, mentioned in latest small cross-sectional research in Norway [139].
Cobalamin (B12)
Numerous biological processes, including DNA methylation and the synthesis of DNA nucleotides, depend on vitamin B12. The body can utilize vitamin B12 in two different ways: as methylcobalamin or as five deoxyadenosine cobalamins. Methylcobalamin is required for the enzyme methionine synthase as a cofactor. Typically, this enzyme is involved in the transformation of the amino acid Hcy into methionine, which is then necessary for DNA methylation [140]. The enzyme that changes l methyl malonyl CoA into succinyl CoA needs the cofactor 5 deoxyadenosyl cobalamin. At this stage, energy is extracted from proteins and lipids [140]. Low levels of circulating B12 are considered to be 65.87ng/mL in the clinic [141]. Shortage may cause cardiovascular disease, anaemia, and neurological issues. The literature is divided on the question of whether a B12 dearth increases the risk of AD. According to one observational study [142], people with AD or cognitive impairment have decreased levels of vitamin B12. On the other hand, employing their phenotypic database, a sizable meta-analysis of B12 conducted in an Icelandic community failed to discover a link with AD [143]. Moreover, two recent Mendelian randomization studies also failed to discover a connection between the two qualities [144. 145].
Lack of vitamin B12 is linked to impairment to the white matter of the brain and spinal cord, which negatively impacts neuronal activity. Unknown is the precise mechanism causing this relationship. Myelin damage brought on by improper methylation of the myelin basic protein (MBP) has been proposed as the underlying mechanism. One-third of myelin protein is made up of MBP, and demyelination in the presence of vitamin B12 shortage may account for several neurologic symptoms, including the correlation with cognitive decline [146].
AD has no available curative treatments at the moment. Cholinesterase inhibitors (ChEIs), which slow the progression of cognitive loss, are the most effective symptomatic pharmaceutical treatment for AD [147]. The response of ChEI treatment varies, nevertheless. Greater responses to ChEI were linked with higher cognitive profiles, prior intellectual employment, healthier lifestyles, marriage, and not living alone, a higher degree of autonomy, and less baseline brain atrophy [147]. The Taiwan National Health Insurance guidelines state that only AD patients whose vitamin B12 inadequacy has been ruled out are eligible for insurance coverage of ChEI treatment. Hence, all individuals with approval for the ChEI prescription must have access to baseline serum vitamin B12 level tests.
Thiamine (B1)
Transketolase, pyruvate dehydrogenase, a-ketoglutarate dehydrogenase, and branched chain a-ketoacid dehydrogenase are enzymes that depend on vitamin B1 and are essential for glycolysis and the Krebs cycle; inadequacies in their activity may lead to reduced glucose metabolism, as seen in the brains of dementia subjects [148]. Memory impairments, neuritic plaques, and τ-hyperphosphorylation are seen in human B1 deficiency (Korsakoff syndrome) in preclinical B1 deficiency animals [148]. Benfotiamine, a more accessible derivative of vitamin B1, was also found to improve spatial memory in APP/PSEN1mutant mice and decrease amyloid plaque and phosphorylated-τ in their brains [149]. The blood B1 diphosphate level was also substantially lower in AD patients compared to controls in a recent case-control study of people, with a sensitivity of 78.1%, specificity of 77.2%, and area under the receiver operating characteristic curve of 77.4% [150]. According to a subsequent study, B1 phosphatases are elevated in the blood of AD patients, which may account for the low B1 diphosphate content in AD [151].
However, it is still unknown how B1 deficiency leads to neurodegeneration. In the brain, B1 deficiency can lead to oxidative stress, endoplasmic reticulum stress, and autophagy, all of which contribute to B1deficiency-mediated neurodegeneration, according to a substantial body of recent study findings [152]. Consequently, treatment approaches that address oxidative stress, endoplasmic reticulum, and autophagy all at once may produce better results. There isn’t enough research in this area, though, to definitively say whether B1 is helpful for AD sufferers.
Riboflavin (B2)
As a cofactor for several flavoprotein enzyme activities, this vitamin participates in a broad range of metabolic routes and functions. Flavin mononucleotide and flavin adenine dinucleotide are its functional forms. According to the available research, vitamin B2 is an antioxidant that may reduce lipid peroxidation and oxidative damage [153]. Vitamin B2 can efficiently raise the level of reduced glutathione (GSH) in tissues since the reduction of oxidized glutathione is a B2 dependent process [153]. The function of antioxidant enzymes is also correlated with a low B2 status, and B2 treatment has been shown to increase activities of antioxidant enzymes [153].
Extensive researches have documented how B2 affects cognitive impairment [154,155]. In a trial from Japan, the prevalence of cognitive decline in older males was substantially higher in the group with a low dietary ingestion of B2 (≤0.96 mg/day) compared to the group with a high dietary consumption of B2 (≥1.11mg/day) [155]. With the exception of its anti-oxidation process, the B2 vitamin may potentially have additional physiological mechanisms that enhance cognitive performance in middle-aged and older people. Another study showed that B2 partially protects against neurological illnesses by promoting anti-oxidation, myelination, and mitochondrial activity [153].
Most recently trial revealed that dietary B2 was preventive for both worldwide cognitive performance and the verbal working memory area [156]. When several variables were taken into consideration during sensitivity analyses, the protective benefits of B2 on cognitive performance were constant and dependable. Neuropsychological test scores throughout the follow-up demonstrated a decreased deterioration in the high-B2 group. Similar to this, B2 administration greatly increased the number of crossings, the time spent in the target quadrant, and the escape delay time (p<0.01) in APP/PS1 mice [157]. B2 dramatically decreased the concentrations of reactive oxygen species (ROS) and methylene-dioxy-amphetamine (MDA). Furthermore, compared to the control group, it led to statistical considerable (p<0.01) elevations in the brain activities of catalase, superoxide dismutase (SOD), and glutathione peroxidase (GSH-Px) in APP/PS1 mice [157]. B2 increased Nrf2 expression while decreasing Keap1 expression in transgenic mice's brain tissue [157]. This study concluded that B2 shields the brain against ROS-induced AD damage, probably because it may be an antioxidant property and because it activates the Nrf2 mechanism. But the precise method by which B2 functions as a neuroprotective factor is still unknown. Research into these distinct physiological pathways of B2 may be necessary in the future.
Niacin (B3)
The term "vitamin B3" refers to two vitamins: nicotinic acid (pyridine-3-carboxylic acid) and nicotinamide (nicotinic acid amide), which are the precursors of the biologically active coenzymes nicotinamide adenine dinucleotide (NAD) and its phosphate analogue, nicotinamide adenine dinucleotide phosphate (NADP). Through its vital participation as a cofactor in the TCA cycle for transformation of acetyl-CoA produced from proteins, lipids, and carbohydrates to ATP, it plays a significant role in the metabolism of carbohydrates and energy.
In a mouse model of stroke, Niaspan (prolonged-release B3 formulation) encouraged synaptic plasticity and axon development [158]. The relative risk (95% CI) of AD in those in the second, third, fourth, and fifth quintiles of B3 ingestion was 0.3 (0.1- 0.6), 0.6 (0.3-1.3), and 0.3 (0.1-0.7), respectively, when compared to those in the first quintile of consumption throughout a 6-year follow-up, and cognitive decline was reduced with higher B3 consumption, which is further support for these possible neuroprotective effects of B3 [159]. Prospective population-based research (the Chicago health and ageing project research) observed an adverse correlation between AD and B3 consumptions after adjusting for a number of dietary (antioxidant nutrients, fats, and vitamins (B1 B2, B6, B9, and B12) and non-dietary (age, education, race, and APOEε4) risk factors for dementia. This suggests that dietary B3 may shield against AD and age-related cognitive decline [159]. Vitamin B3 (particularly nicotinamide) could be useful for AD although the epidemiologic evidence is still sparse and inconsistent, especially in light of the fact that it mediates important biological pathways (like, energy metabolism, mitochondrial functions, calcium homeostasis, survival and cell death). NAD+ contains life-extending properties, which are crucial for brain activities like neurotransmission, memory, and learning. Since NAD+ reduction and mitochondrial impairment are essential for synaptic plasticity and are typically present in ageing and AD onset [160], boosting NAD+ brain levels in AD mouse models can reverse mitochondrial dysfunction and prevent cognitive loss [160].
Both in-vitro (organotypic hippocampal slice cultures) and in-vivo (AD model rats) trials have highlighted the defensive effects of vitamin B3 against A-beta-induced neurotoxicity. Nicotinamide or nicotinamide mononucleotide also mitigate amyloid toxicity, through minimizing appearance of AD-related genes (amyloid precursor protein and presenilin 1) and ROS generation, then by enhancing neuron survival [161,162]. Variations in NAD+ availability can lessen AD pathogenesis through modifying SIRT1 function as well as delaying ageing and diseases linked to ageing [163]. In particular, deacetylase activity of SIRT1 has been demonstrated to assist the non-amyloidogenic route of AD and to reverse circumstances, such as neuroinflammation, oxidative stress, and mitochondrial dysfunction, having contributed to and exacerbating AD. Numerous research findings have highlighted the critical role of SIRTs in AD prevention [164].
Pantothenic acid (B5)
The B5 vitamin, a coenzyme A (CoA) precursor, is essential for the metabolism of fatty acids and carboxylic acids in a variety of species [165]. Above its function in oxidative metabolism, CoA participates in the synthesis of cholesterol, amino acids, phospholipids, and fatty acids, which helps to support the structure and operation of brain cells [165]. Importantly, this vitamin functions in mechanisms involving CoA in the production of several neurotransmitters as well as steroid hormones. Rapid reactive proteins associated with inflammation, adhesion molecules, and pro-inflammatory cytokines are examples of biological defense resources that may be inappropriately consumed as a result of higher CoA activity brought on by excessive B5 intake. But additional research is needed to confirm this idea.
In 2018, researchers looked examined the correlation between vitamin consumption and cerebral A-beta load in AD patients with cognitive impairment [166]. Brain MRI and 18F-florbetaben positron emission tomography were performed on all individuals. The 15 vitamins' dietary consumption was assessed using the Food Frequency Questionnaire. The A-beta-positive group consumed more vitamin A (p = 0.063), K (p = 0.042), beta-carotene (p = 0.081), B2 (p = 0.063), B3 (p = 0.097), B5 (p = 0.092), and B6 (p = 0.027) than the A-beta-negative group. Vitamin B5 consumption was identified as an independent predictor of cerebral A-beta load by multivariate linear regression analysis (beta = 0.287, p = 0.029) [166]. The findings suggested that people with cognitive impairment may have higher cerebral A-beta burdens after consuming B5. These findings might shed light on prospective AD preventative tactics.
Pyridoxic acid (B6)
Pyridoxine, Pyridoxal, and Pyridoxamine are the three physiologically comparable molecules that comprise vitamin B6. The first (Pyridoxine) is abundantly present in plant materials, whereas the other two are present in animal tissues. Because vitamin B6 is essential for the production of various neurotransmitters, such as dopamine, serotonin, norepinephrine, and gamma-aminobutyric acid, having a vitamin B6 shortage has a major impact on brain activity [167]. The development of several cognitive functions may be related to the absence of these neurotransmitters. The Hcy may have directly damaging effects on CNS neurons in addition to being a risk factor for cerebrovascular disease. Numerous investigations have suggested that AD’s etiology includes hyperhomocysteinemia [168,169]. Results from observational research have shown that numerous B-vitamins, particularly vitamin B6, have an impact on plasma Hcy concentrations [129,170]. According to a study, persons with AD have lower plasma levels of pyridoxal-5-phosphate compared to healthy controls [171]. There is no proof that vitamin B6 addition affects older people's cognitive abilities in any way. Indicators of vitamin B6 availability in older men's biochemical profiles are improved by vitamin B6 treatment, indicating that some may lack the vitamin. Additional randomized controlled trials are required to assess the effects of vitamin B6 on lowering the risk of general population cognitive decline and on enhancing cognitive and emotional functions in dementia patients. Experiments must last for a long time and be set up to allow subgroup analysis limited to people with biochemical signs of vitamin B6 insufficiency.
Biotin (B7)
It is also called the anti-egg white damage factor or vitamin H. Long-term use of raw egg whites might cause a B7 shortage. Consuming raw egg whites results in a deficiency because avidin, a glycoprotein that binds B7 with a high affinity, is present. In processes mediated through five carboxylase enzymes concerned in the TCA and the FAs metabolism, vitamin B7 serves as a coenzyme: propionyl-CoA carboxylase, 3-methylcrotonyl-CoA carboxylase, pyruvate carboxylase, and acetyl-CoA carboxylases 1 and 2 [172]. Vitamin B7's involvement in epigenetic regulation through histone biotinylation, gene expression, and cellular communication has recently been expanded [173]. Vitamin B7 specifically has been reported to increase soluble guanylate cyclase (sGC), increasing levels of cyclic guanosine monophosphate (cGMP), and influence the production of enzymes involved in the homeostasis of glucose, such as glucokinase and phosphoenolpyruvate carboxykinase [172].
High-dose of B7 supplement (100 mg, 3times daily) was found to have a stabilizing effect on the course of multiple sclerosis (MS) in a current controlled experiment [173]. A case may be made for direct stimulation of soluble sGC through pharmacological doses of B7 plays an important function in this regard even though this effect has been referred to an improvement of B7 necessary cofactor role in the brain. The effectiveness of high-dose B7 in MS may be due to cGMP's anti-inflammatory effects on the cerebral microvasculature, oligodendrocyte development, and the synthesis of neurotrophic factors by Schwann cells that are considered to be useful in treating MS [173]. However, B7 may have more neuroprotective benefits due to its capacity to increase cGMP production in the brain. The cGMP has neurotrophic-mimetic effects in a variety of neurons and brain cells, activating the PI3K-Akt and Ras-ERK pathways in the process to support neuron viability and plasticity [173]. Agents that have a compensatory impact by increasing cGMP levels tend to restore effective long-term potentiating in mouse models of AD. In AD, A-beta decreases this mechanism by reducing sGC activity [174]. These results imply that high-dose B7 may be useful for the prevention and treatment of AD, together with cGMP's capacity to prevent neuron death. In addition to promoting neurogenesis, cGMP also inhibits atherogenesis and hypertrophic remodeling in the cerebral vasculature, which may reduce the risk of stroke. Concurrent treatment of brain-permeable phosphodiesterase-5 inhibitors may enhance the neuroprotective efficacy of large doses of B7 [174].
However, attention in vitamin B7's neuroprotective activity is recent, and more research investigating potential pathways and modes of action in-vitro as well as in-vivo is required to make clear its function in the brain. Vitamin B7 may be involved in and improve a number of acute and chronic neurological diseases [175].
Folic acid (B9)
Folate is essential for the CNS's development, differentiation, and regeneration [176]. Vitamin B9 is a necessary nutrient that contributes a methyl group to a number of metabolic pathways, including the production of neurotransmitters, DNA, the regulation of gene expression, the synthesis and metabolism of amino acids, and the manufacture and repair of myelin [176].
The supplementation of vitamin B9 can prevent different diseases of the CNS, like AD, neural tube defects and development delays [176]. This vitamin is vital for the neurological system at all ages and is extremely critical in the form of methyl folate, which may be partly responsible for the multiple CNS methylation processes. Many doctors advise AD patients to take vitamin B9 and other B vitamin supplements to improve cognitive function, but their efficacy is still debatable [136]. It was noticed that people with low serum levels of vitamin B9 are more likely to develop AD [177]. In a similar trial [178], high dietary B9 intake (top quintile: median, 742 µg/day) was associated with a faster rate of cognitive decline than that felt by those in the lowest intake quintile (median, 186 µg/day), which was conducted prospectively on older adults 65 years of age or older living in the community. Supplementation of B9 >400 µg/day was likewise associated with higher rates of decline.
There are many possible processes that link vitamin B9 intake to the emergence of AD, some of which may be connected to Hcy. Dementia and cognitive decline are both associated with serum Hcy concentrations. Numerous reasonable mechanisms by which high Hcy amounts might elevate the risk of dementia have been speculated (e.g., an impact on cerebrovascular pathology, direct neurotoxic effects, or an effect on the neurofibrillary tangle burden and A-beta accumulation via impacts on methylation reactions) based on the recognized effects of B9 and B12 inadequacies and anomalous Hcy metabolic activity on the formation and maintenance of the nervous system [179]. None of these methods, therefore, has been categorically demonstrated. Because vitamin B9 administration lowers serum Hcy amounts and could consequently be a way to assist combat dementia or age-related cognitive decline, addressing the question is crucial from a practical standpoint. The effectiveness of B9 therapies in mitigating cognitive decline has been investigated in randomized studies; however the findings are inconclusive [180,181]. Low B9 levels have been proven to cause an inflammatory state in epidemiological investigations, which may help to explain the link between vitamin B shortage and the risk of AD [179].
The patients with neuropathologically diagnosed AD and no major cerebrovascular disease or atherosclerosis have been demonstrated to be at risk due to the Hcy [182]. Give proof from an animal study suggesting B9 deficit and Hcy could be strongly associated to amyloid toxicity through preventing neurons from repairing DNA, which makes them more susceptible to oxidative damage brought on by A-beta [183]. This implies a non-vascular mechanism via which B9 consumption may be linked to the onset of AD. There are facts that Hcy can induce direct damage to brain cells, which supports another nonvascular mechanism [169]. The link between B9 consumption and the development of AD has also been related to non-Hcy processes involving methylation actions in the brain [184]. Regardless of age and educational level, a study revealed drastically reduced B9 levels in AD patients compared to MCI subjects and healthy older persons [185]. There were no disparities between MCI participants and old controls. Additionally, lower B9 levels were associated to decrease cognitive functioning, especially in terms of memory, language, and psychomotor speed [185]. The median readings were reported to be greater than the lower range of laboratorial reference standards (i.e. 4.2 to 19.9 ng/ml), despite reduced B9 levels in AD patients [185].
Ascorbic acid (vitamin C)
The water-soluble antioxidant vitamin C, which is found in the cytosol and quenches many radical types, can replenish other antioxidants like glutathione and vitamin E, and it is particularly abundant in the brain [186]. The capacity of vitamin C to give electrons (as a reducing agent) is the basis for all of its physiological and biochemical activities. Ascorbic acid passes through two successive, reversible oxidations: after losing the first electron, it produces the ascorbate free radical as an intermediate product, which is then changed into dehydroascorbate after losing the second electron. Vitamin C is a potent antioxidant and free radical scavenger at physiological quantities in plasma and several organs, including the CNS [187].
Studies conducted in both in-vivo and in-vitro showed that vitamin C decreased oxidative stress and the development of A-beta-oligomers [188]. Cohort study results on the impact of vitamin C supplements on AD are inconclusive. There was no difference in the prevalence of AD during the 4-year follow-up in prospective research (n=980) assessing the connection between 4 years of vitamin C and vitamin E intakes and the risk of AD [189], as well as similar correlation was also revealed in another prospective trial (n = 5395), demonstrating that vitamin C consumption was not connected to an increased risk of AD [96]. But findings from largely published prospective observational research with a sample size of 4740 people revealed that taking vitamins C and E together for at least three years was related to a decrease in the occurrence and incidence rate of AD [190]. Generally, there is overwhelming proof in the literature that preserving adequate vitamin C levels can have a protective effect against AD, although preventing vitamin C deficiency is probably more advantageous than adding supplements to a regular, healthy food [191].
In particular, the scavenging activity against ROS, the control of neuroinflammation, the suppression of the fibrillation of A-beta peptide, and the chelation of Fe, Cu, and Zn are the key mechanisms linked to vitamin C neuroprotection [192]. According to recommendations, vitamin C contributes significantly to the pathophysiology of AD by directly protecting neurons from oxidative stress. It has been well-established that neurodegeneration is caused by a disproportion in vitamin C homeostasis [193]. This vitamin serves as a crucial CNS antioxidant, releasing from glial cells to the synaptic cleft and being taken up by neurons as an antioxidant defense to sustain synaptic activity and neuronal metabolism [193]. It was shown that the astrocyte-neuron contact serves as an important mechanism for recycling vitamin C, contributing to the brain’s antioxidative defense [194]. In reality, vitamin C helps to regenerate endogenous antioxidants (GSH, catalase, vitamin E) and is a first-line antioxidant defense against ROS reactivity [194].
Multiple cross-sectional investigations have shown that AD patients' CSF-to-plasma vitamin C ratios are lower than those of controls. Recent research in particular revealed that blood-brain barrier (BBB) damage negatively affects the ratio of CSF to plasma vitamin C, which is crucial for avoiding cognitive decline in AD [195]. It is yet unclear, though, whether the dysfunction of the vitamin C transports carrier, or the compromised integrity of the BBB is to blame [195]. Moreover, four of the eleven studies that looked at the connection between CSF and plasma vitamin C levels and cognitive decline (including AD) also looked at the ratios of CSF to plasma vitamin C. According to one study’s findings, AD patients had considerably lower mean CSF-to-plasma vitamin C ratios than controls [196]. Final results were not reached from a second prospective examination of CSF vitamin C, rates of cognitive decline, and BBB, however a larger CSF-to-plasma vitamin C ratio was associated to a slower rate of decline [186].
In both European and American nations, eight sizable population studies have examined the connection between vitamin C consumption and AD-type dementia [186]. The neuroprotection related to vitamin C has not yet been identified, though. Another significant population study revealed that vitamin C plays a preventive function in people with vascular dementia and cognitively healthy individuals, although AD was not one of these conditions [197]. In a population study of North Carolina, 616 seniors over 75 years who had used vitamin C supplements for a long time did not show any neuroprotection against developing AD [198].
The clinical outcomes may need to be interpreted within a set of constraints. The observational research on vitamin C consumption, plasma concentrations, and its advantages for ageing and cognition are highly inconsistent. It is notable that many clinical studies typically did not include older individuals with polypharmacy or comorbidities. The populations’ majority at risk for vitamin C shortage are excluded by these exclusion criteria, which seriously impair the validity of the results. Harrison and coworkers [191] summarized the classification of groups according to vitamin C level varies significantly among research [191]. Insufficient concentrations of vitamin C are lower than 1.27 mg/L, suboptimal levels are in the range of 1.27 to 4.37 mg/L, adequate plasmatic levels are above 3.22 mg/L, and optimal levels are in the range of 5.76 to 6.91 mg/L [191]. Another source of variation is the wide range of vitamin C pills, which go from 27 to 230-270 mg/day. Last but not least, if vitamin C consumption did not exceed 500 mg per day, it was linked to improved cognition; higher plasmatic levels (1.0 g per day) were allied to worse cognitive function [191].
Additionally, a high vitamin C level may be harmful to patients because it encourages the Fenton reaction (a reaction that creates hydroxyl radicals in Fe rich tissues like brain or spinal cord white matter). High amounts of antioxidants, such as vitamin C, might speed up this reaction and, as a result, may worsen the inflammatory state because of radical formation. Ferrous (reduced form of iron ion) is a component of this reaction [129]. Reliable plasmatic measurements with many measurement points are needed to fully understand the beneficial effects of vitamin C. The dependability of the results may be impacted by inaccurate study designs and techniques [186]. The accuracy of results is also impacted by the absence of standardization between the use of single nutrients or multivitamins and the lack of systematic monitoring of plasmatic concentrations of vitamin. It is essential to establish the distinction between dietary intake and manufactured supplements, as well as the average of their bioavailability. The concentrations of vitamin C necessary to positively alter brain ageing are mostly unknown as of right now. There are still questions about two major issues, including whether there is a connection between a vitamin C deficit and cognitive decline (including dementia). It has not yet been determined if ascorbic acid insufficiency is a cause of AD neurodegeneration or only an epiphenomenal factor. Further research into vitamin C’s involvement in supporting brain function and healthy ageing is made possible by its powerful free radical scavenging characteristics, well-characterized transport kinetics, and excellent bioavailability in the CNS. A MEGA-PRESS mediated spectrum can be used to find the contents of vitamin C in the brain (MEGA-PRESS, MEGA-point-resolved spectroscopy) [199]. The study found a link between blood and brain amounts of vitamin C and offers a fresh conceptual framework for future research examining vitamin C's function in the brain. Hence, all of these studies’ direction may help to clarify how vitamin C affects brain ageing in the future and, ideally, offer a fresh conceptualization of AD.
Quasi vitamins
A number of other factors appear to satisfy the criteria of vitamin status under only certain conditions. These are recognized with the status of “quasi-vitamin”. Details of a function for “quasi-vitamins” in providing shield against AD are given below:
Choline
A component similar to vitamin B called choline (2-hydroxy-N,N,Trimethylethanaminium) is present in many everyday meals and is crucial for a number of cell processes. Methionine, the B vitamins B9, B6, and B12, and choline are all tightly connected in terms of metabolism, therefore modifications in one of these metabolic pathways will also affect the others [200]. It primarily occurs naturally as phosphatidylcholine (lecithin), which has the potential to penetrate the BBB [9]. Evidence suggests that specific choline-containing phospholipids, such as Cytidine 5′-diphosphocholine and choline alphoscerate, improved the cognitive abilities of patients with neurodegenerative diseases. It serves as a precursor of the neurotransmitter acetylcholine, a methyl donor in one-carbon metabolism, and as an essential component of membrane phospholipids [201]. There are at least two ways that choline protects the brain from AD [202]. First, choline prevents the development of amyloid-beta plaques, the pathology that characterises AD. Secondly, supplementation of choline lessens microglia activation. Overactive microglia produce inflammation in the brain and may eventually result in nerve cell loss, impairing cognitive function.
It is noteworthy that acetylcholine causes microglia to express alpha7 nicotinic acetylcholine receptors (α7nAchR) [203]. Immune cells that live in the brain, called microglia, carry out maintenance tasks that are crucial for the health of neurons [204]. Though, constantly active microglia, which is more prevalent in AD, cause chronic brain inflammation that eventually results in neuronal death [205]. In the APP/PS1 mouse model of AD, there is a rise in activated microglia, and this is associated with an upregulation of the α7nAchR gene [206]. Acetylcholine levels have been found to be lower in the brains of AD patients. As a result, raising acetylcholine might cause α7nAchR to activate, which might lead to a reduction in active microglia [206]. The majority of recent studies [207] demonstrated that choline can act as an agonist for the Sigma-1 receptor (σ1R), which is essential for controlling brain inflammation. Microglias express σ1Rs, which are similar to α7nAchRs. When these receptors are dysregulated, microglias are activated and release pro-inflammatory chemicals [208]. Indeed, in vitro research has demonstrated that σ1R agonists reduce microglia activity [208]. Therefore, improving the activity of σ1Rs may lessen AD-related brain inflammation and neurodegeneration.
According to the US Standing Committee on the Scientific Evaluation, adult women (>19 years of age) should consume 425 mg of choline per day, while adult men should consume 550 mg per day [209]. Average daily intake of choline in human on an ad libitum diet was noted to be lower than that of recommended daily intake, particularly in women [210]. Facts also revealed that even the existing recommended daily intake could not be ideal for a healthy ageing process [211]. This suggested that adding more choline to your diet might help avoid neuropathological variations connected to ageing of the brain. For adult females and males (>19 years of age), the tolerable upper limit of choline that is unlikely to result in side effects is 3,500 mg/day, which is 8.24 times higher than the 425 mg/day advice for females and 6.36 times higher than the 550 mg/day prescription for males [209].
The researchers most recently investigated whether dietary supplementation of choline over the course of a person's life could lessen AD-like pathology and improve memory problems in the APP/PS1 mice model of AD (210). From 2.5 to 10 months of age, they subjected female APP/PS1 and NonTg mice to either a control choline diet (1.1 g/kg choline chloride) or a choline-supplemented diet (5.0g/kg choline chloride). The spatial memory of mice was evaluated in the Morris water maze before a neuropathological analysis. In APP/PS1 mice, lifelong choline administration substantially decreased A-beta plaque load and enhanced spatial memory [210]. The amyloidogenic processing of APP was reduced, disease-related microglial activation was decreased, and the α7nAch and σ1Rs were down-regulated as a result of these modifications [210]. The findings demonstrated that simple dietary changes could have a significant impact on reducing AD pathology and that lifelong choline addition generated significant benefits.
A key intracellular precursor of the phospholipid phosphatidylcholine is citicoline (cytidine-5'-diphosphocholine). The majority of clinical trials using dietary citicoline for AD subjects revealed that treated patients improved their attention and memory and scored better on cognitive evaluation scales compared to control groups [212,213,214]. A frequent choline molecule and acetylcholine precursor in the brain, choline alfoscerate (l-a glycerylphosphorylcholine, or a-GPC) has been proven to be beneficial in the treatment of AD and dementia. In regard to choline alfoscerate, a double-blind RCT displayed that treatment with 400mg of choline alfoscerate 3 times per day substantially enhanced scores on the Mini-Mental State Examination, the Global Deterioration Scale, the AD Assessment Scale (Behavioral and Total), as well as the Global Impression Scale after 90 days among patients with mild-moderate AD, whereas the scores in the placebo group stayed largely the same or got worse [215]. Similar good results with choline alfoscerate addition for AD individuals have been documented in numerous other investigations [216,217]. A review of clinical trials of cholinergic precursors revealed that supplementing with lecithin or phosphatidylserine improved subjective memory but did not ultimately improve outcomes for AD patients’ QoL, cognitive function, or mortality. This suggests that not all cholinergic precursors have been proven to be effective treatments for AD [218]. The causes of this antecedent treatment strategy’s lack of effectiveness are yet unknown. Despite extensive research, not all elements of choline deposition, metabolism, and use in the production of acetylcholine have been fully understood [216]. Therefore, it seems that various cholinergic precursors have a variety of effects, and more research is required to investigate these precursors in order to develop appropriate treatments for AD patients.
Inositol
Inositols (1,2,3,4,5,6-cyclohexanehexol) are polyols with six-carbon rings that have been hydroxylated on each carbon. There are several of these sugar-alcohol isomers that are physiologically active, the most prevalent of which being myo- and scyllo-inositol [219]. Through active transport, myo- and scyllo-inositol concentrations in the brain are 100 times higher than those in the periphery [219]. Two sodium/myo-inositol transporters (SMIT1, SMIT2) may be in charge of controlling the levels of inositol in the brain. These inositols have the ability to block beta-secretase-1, which is necessary for converting APP into A-beta or A-beta oligomerization [205]. Individuals who have MCI are more likely to develop AD. Whereas myo-inositol concentrations in AD were elevated throughout the white matter, they were elevated in the parietal white matter in MCI [220]. As a result, the myo-inositol concentration in MCI may be considered a precursor to AD [220]. Human clinical trials using scyllo-inositol are now being conducted to treat people with mild to moderate AD. Cognitive impairments and AD-like amyloid pathology are successfully treated with scyllo-inositol in TgCRND8 mice [221]. TgCRND8 animals exhibit several of the hallmarks of AD, such as plaque formation, elevated cerebral A-beta levels, A-beta aggregation, and cognitive deficits as the disease advances [221]. Treatment with scyllo-inositol greatly increased survival rates, synaptic function, and spatial memory in TgCRND8 mice [222]. These advantageous outcomes happened in rats administered scyllo-inositol both therapeutically and prophylactically, before symptoms became clearly obvious [222].
A study revealed that decreased emergence of affective neuropsychiatric symptoms in AD was related with particular scyllo- and myo-inositol variations in brain evaluated through magnetic resonance spectroscopy [223]. Brain myo-inositol modulation may serve as a partial mediator of the neuropsychiatric effects of ELND005 (scyllo-inositol) taken orally. Scyllo-inositol levels in the brain increased with ELND005 administration at doses of 250 mg and 1000 mg, reaching apparent saturation above 1000 mg [224]. The percentage varies from baseline at the 250 mg and 1000 mg doses were -36% to -45% and -61% to -64%, respectively, and also reached apparent saturation at doses over 1000 mg. The brain levels of myo-inositol demonstrated a reciprocal reduction at the same time points [224]. From 24 to 78 weeks, these effects on scyllo- and myo-inositol were noted, and they were comparable in mild and moderate individuals. At any given visit, total brain inositol concentrations were typically steady, and the negative correlation between scyllo- and myo-inositol (at 24, 48 wks: r= 0.86, 0.9, both p 0.0001) was strong [224].
Carnitine (beta-hydroxy-gamma-N-trimethylaminobutyric acid)
Lysine and methionine, two necessary amino acids, are used to create carnitine, a branched non-essential amino acid. Carnitine is a nutrient that may be received from food and is also produced in the kidney, liver, and brain. Under some conditions, such as when intracellular concentrations are low, it is regarded to be "conditionally essential" for human (e.g. elderly patients, premature infants, diabetes and genetic conditions resultant in primary or secondary carnitine deficiency) [225]. Well-known cofactors include the vitamins C, B3, B6, and ferrous iron, and deficits of any one of these can cause a carnitine shortage [226]. Main roles to transfer acetyl groups from FAs into mitochondria for ATP production, has function in acetylcholine production, and documented curative benefits on mitochondrial role, physical activity, as well as remembrance in older rats [227]. Both free carnitine and its acyl derivatives can be found in cells and tissues. Acetyl-L-carnitine (ALC) is produced by enzymes called carnitine acyl-transferases [227].
ALC may have a positive impact on dementia, cognition, and behavior in elderly and AD people, according to several investigations [228,229,230]. ALC may work in AD by a variety of methods, including the repair of cell membranes and synaptic function, stimulation of cholinergic activity, restoration of brain energy, defense against toxins, and neurotrophic effects through the stimulation of nerve growth factor and protein acetylation [231]. These conclusions, though, have not been supported by all studies [232]. Scientists noticed in a meta-analysis research involving 16 trials that there were significant methodological and evaluation tool differences between early and later investigations, making it impossible to compare them [229]. ALC also had statistically important treatment effects for clinical global impression at 12 and 24 weeks, as well as for the Mini Mental State Examination (MMSE) at 24 weeks, according to the results of meta-analysis [229]. Another meta-analysis investigation reported similar clinical global impression and many psychometric test substantial treatment effects of ALC [233]. Researchers measured the concentrations of free L-carnitine, ALC, and acyl-L-carnitine in the serum of healthy individuals, AD patients, and patients of AD at risk [230]. ALC levels gradually dropped from the group of healthy patients (0.06 mg/dL) to those with subjective memory complaints (0.049 mg/dL), MCI (0.045 mg/dL), and finally those with AD (0.040 mg/dL; p<0.001) [230]. When comparing patients with AD or at risk for AD to healthy people, other acyl-carnitines also displayed a similar downward tendency [230]. The serums levels of ALC and other acyl-L-carnitines may be used to assist distinguish between patients who are at risk for developing AD and those who would benefit from therapy with ALC. Before use in clinical practice, additional verification on a larger number of samples in longitudinal research is required. Recent research by a team of experts found that giving dementia patients with vascular cognitive impairment ALC 1,500 mg/day resulted in significant improvements on the Korean version of the Montreal Cognitive Assessment [234]. The population in this study tolerated ALC well as well. Despite the study's methodology and sample size limitations, this research revealed that using ALC with dementia patients may have some advantages. It is advised to do a sizable investigation with patients who have never taken cholinergic drugs in order to further confirm these findings.
Mechanisms related with Alzheimer’s disease risk
Numerous biologically conceivable methods have been proposed to explain the function of vitamins in AD, despite the lack of a clearly defined mechanism. The several vitamins (as indicated in Figure 2) that are needed as cofactors in these pathways or that affect the severity of brain illness, as well as the six metabolic pathways or pathologies associated with an increased risk of AD [235].
1) One-carbon metabolism in AD
As co-factors for one-carbon transfer reactions, which are essential for the synthesis of DNA and RNA nucleotides, the metabolism of amino acids, and the maintenance of methylation reactions, vitamins B2, B6, B9, B12, and choline are needed in the brain [236]. Therefore, the postulated processes by which low levels of or a shortage in B vitamins may increase the risk of AD entail disruption of the intricate regulatory network sustaining one-carbon metabolism [237]. This may lead to increased uracil misincorporation into DNA, hyperhomocysteinemia and/or decreased methylation status in the brain tissue, as well as changes to RNA and neurotransmitter products [238]. The immune system may be affected by higher Hcy and/or decreased B-vitamin levels, which could lead to more inflammation and antioxidant damage [239]. Rats lacking in B vitamins showed cognitive impairments and AD, which were linked to aberrant methylation [238]. Improved B-vitamin status may assist to mitigate this risk. Certain genetic polymorphisms, such as the prevalent MTHFR 677C/T polymorphism, can interfere with normal one-carbon metabolism and have an effect on AD, independent of Hcy [239]. The B-vitamins particularly B6, B9, and B12 act in concert to support a variety of biochemical and molecular processes, including the network of one-carbon metabolism [239].
A dicarboxylic acid called methylmalonic acid (MMA) is involved in the metabolism of a number of branched-chain amino acids and fatty acids [240]. MethylmalonylcoA is converted to succinylcoA with the help of vitamin B12 as a cofactor. When there is a vitamin B12 deficit, MMA CoA is converted to MMA, which causes an intracellular rise in MMA and higher serum concentrations [240]. The most sensitive serum indicators of early intracellular vitamin B12 and B9 insufficiency are recognized to be the Hcy and MMA [241]. The CSF total Hcy and CSF MMA may serve as early indicators of B9 or B12 deficiency functional deficit at the neuronal level. The Hcy and MMA are neurotoxic and may have a role in the development of AD [240].
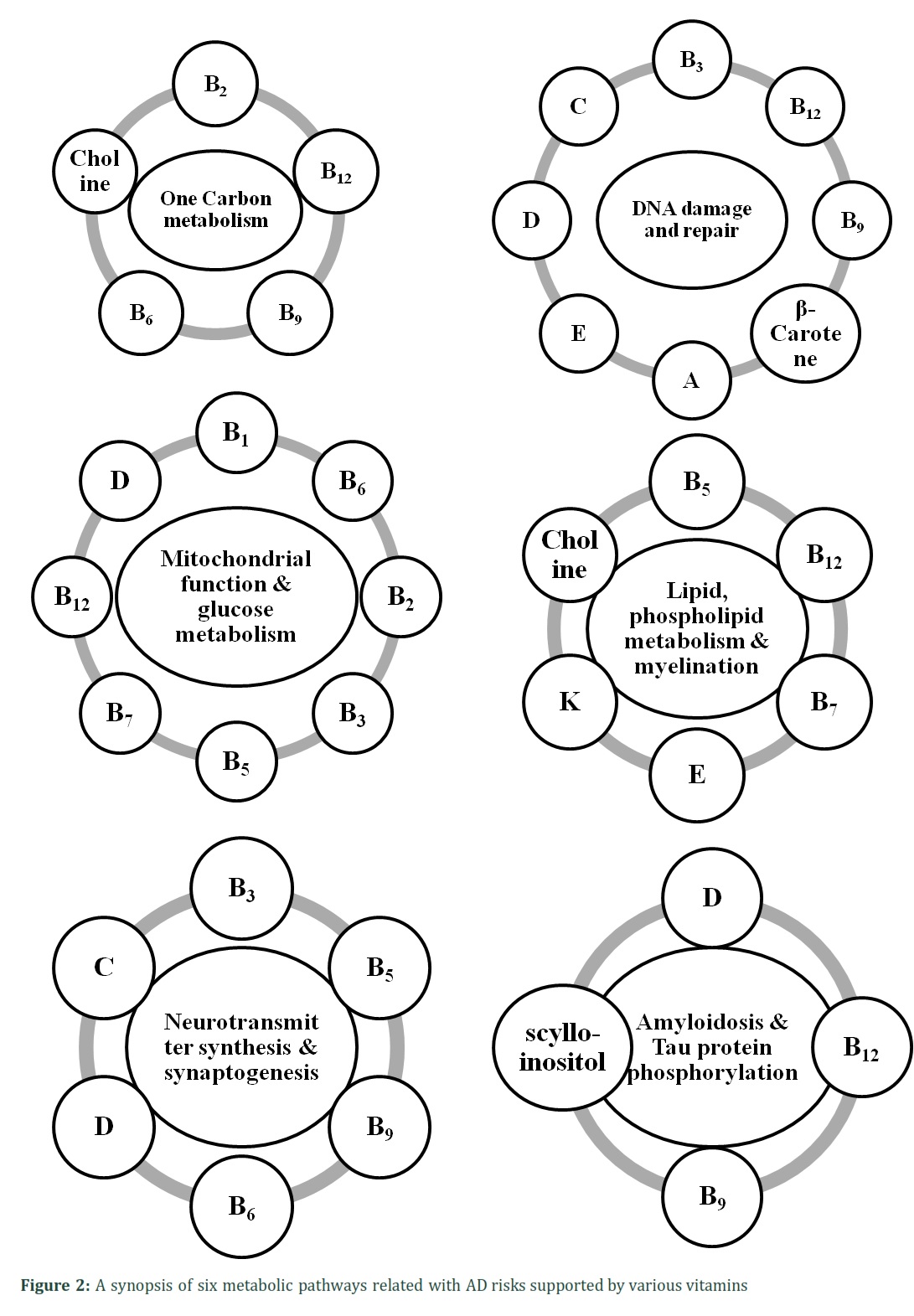
2) DNA Damage and Repair
Radiation, nutrition, and environmental pollutants are examples of exogenous sources of DNA damage. Chemical instability, such as depurination, spontaneous mistakes during DNA replication and repair, and ROS, which are byproducts of normal metabolism, are all examples of endogenous sources of DNA damage [242]. Base excision repair, nucleotide excision repair, mismatch repair, and double-strand break repair are prominent DNA repair processes in mammalian cells [242]. It is recognised that AD is accompanied by DNA damage, such as single- or double-strand breaks (DSBs) [243]. In recent, it was demonstrated that A-beta or A-beta peptides cause oxidative stress, which results in DSBs [244]. It was already known that the vitamin A derivative ATRA had neuroprotective properties against the amyloid cascade. It decreases things like the synthesis of A-beta peptides and their oligomerization [244]. Moreover, RA and its receptor are implicated in the repair of DSBs caused by A-beta as well as in the prevention of A-beta production, A-beta oligomerization, and plaque clearance [244].
Oxidative stress occurs when oxidizing molecules like ROS overwhelm a cell's protection mechanism. Numerous disorders have been suggested to be correlated with ROS, and neurodegenerative conditions like AD and Parkinson's disease appear to be among them [245]. Reactive nitrogen species (RNS) can damage nucleic acids and cause the deamination of the nucleotide bases. Nitrotyrosine is upregulated in response to RNS-mediated DNA damage, and it is also utilized as a marker for this type of damage [246]. Moreover, DNA is also damaged by advanced lipid peroxidation end products such as 4-hydroxy-2-nonenal (HNE), through the formation of DNA-HNE adducts [246].
Biological fluids, peripheral tissues, and brain areas of AD patients have been exposed to have a substantial number of DNA damage markers, notably oxidative DNA damage markers [246]. Antioxidants like vitamins C, E, and beta-carotene can block the harmful effects of hydroxyl radicals, superoxide, and hydroperoxide radical anions [247]. Generally speaking, vitamin C and vitamin E combos are more potent antioxidants than either vitamin alone. In such circumstances, vitamin E can be restored from its oxidatively damaged state by interacting with vitamin C, which is considerably more prevalent in cells [247]. Vitamin D maintains proper mitochondrial functioning, which aids in cellular oxidation and reduction (redox) management [248]. Loss of redox regulation of the cell cycle may cause abnormal cell growth, cell death, and the onset of AD [248]. Increased risks of disease are related to vitamin D insufficiency, as assessed by serum 25(OH)D concentrations of less than 30 ng/mL [248]. Normal serum levels of 1,25(OH)2D and 25(OH)D are necessary for ideal cellular function and act as a preventative measure against excessive oxidative stress-related DNA damage [248].
One of the few vitamins with a direct connection to DNA synthesis, DNA repair, and cell death is vitamin B3. This vitamin is required as a substrate for poly-ADP-ribose polymerase-1 (PARP-1), which is involved in cleavage and rejoining of DNA and in telomere length maintenance [249]. Increased DNA oxidation, DNA breakage, and an enhanced rate of chromosome damage are results of its absence [249]. Such damage appears to have a significant role in the etiology of several disorders, including AD, possibly as a result.
Numerous procedures that affect DNA integrity depend on vitamin B9, including DNA synthesis and methylation. Studies conducted in vitro have demonstrated that a B9 deficiency increases uracil assimilation into the DNA of human lymphocytes in a dose-dependent manner. Vitamin B9 treatment decreases chromosomal breakage and DNA uracil integration in human cells [250]. Low B9 levels have been suggested as a possible AD promoter [177].
Vitamin B12 is crucial for DNA synthesis and maintains the structural integrity of crucial chromosomal areas like centromeres and subtelomeric DNA [251]. Important chromosomal areas, such as the centromeres and the subtelomeric DNA, are protected structurally by vitamin B12, which is necessary for DNA synthesis. Low serum levels of B12, which are caused by inadequate food intake, affect DNA synthesis. Therefore, DNA damage may result from a B12 shortage. Low B12 serum levels were revealed to be a major factor in AD [251]. Owing to their complimentary roles in the "folate" and "methionine" cycles, the roles of the vitamins B9 and B12 are inextricably intertwined. In fact, a lack of vitamin B12 causes a functional lack of B9 because B9 gets stuck in the form of methyltetrahydrofolate [25]. A true or functional B9 deficiency results in decreased DNA stability and repair, gene expression/transcription, and genomic and non-genomic methylation reactions in brain tissue, which may impede neuronal differentiation and repair, encourage hippocampal atrophy, demyelination, and compromise the integrity of membrane phospholipids impairing the propagation of action potentials [25].
3) Mitochondrial Function and Glucose Metabolism
It has been hypothesized that A-beta-induced mitochondrial dysfunction reduces ATP synthesis efficiency and boosts ROS production in AD [252]. Multiple indicators point to mitochondrial dysfunction in AD. First, AD has been associated to impaired energy metabolism in the brain [253]. Reduced neuronal expression of genes that code for mitochondrial electron transport chain subunits was linked to a reduced cerebral glucose metabolism in AD [253]. Second, patients with AD have lower activity levels of critical oxidative metabolism enzymes as pyruvate dehydrogenase complex, a-ketoglutarate dehydrogenase complex, and cytochrome oxidase [253]. Third, A-beta-induced mitochondrial dysfunction exacerbates calcium homeostasis failure [253]. Finally, it has been observed that AD patients have greater mitochondrial DNA oxidative damage, which may result in more mitochondrial DNA mutations [253].
Mitochondrial dysfunction may result from a vitamin D shortage. One of vitamin D's primary roles is to keep the mitochondrial respiratory chain active [254]. The uncoupling protein, which is found on the inner mitochondrial membrane and controls thermogenesis, is likewise regulated by vitamin D [254]. Due to a decrease in the nuclear mRNA molecules and proteins that support mitochondrial respiration during vitamin D deficiency, mitochondrial respiration decreases [255]. Because of a vitamin D-dependent decrease in the expression of complex I of the electron transport chain, ATP synthesis specifically diminishes. As the electron transport chain breaks down, more ROS are produced, which causes oxidative stress and eventually lead to the development of AD [254].
Through their active metabolites, the B vitamins (especially B1, B2, B3, and B5) have a direct impact on mitochondrial aerobic respiration (TCA cycle) and energy production (electron transport chain and ATP formation) [256]. However, the function of vitamin B7 is less clear. Acetyl-CoA and succinyl-CoA, which are secondary byproducts of biotin-dependent processes, can feed into the TCA cycle and so contribute to energy consumption [256]. While vitamin B9 and B12 are necessary for nucleotide biosynthesis and amino acid metabolism, vitamin B6 is necessary for FeS production, de novo synthesis of NAD+, and substrate metabolism [257]. Furthermore, vitamin B12 is as well essential for the mitochondria's production of succinyl-CoA from methylmalonyl-CoA. [257].
4) Lipid and Phospholipid Metabolism and Myelination
Lipids can be broadly divided into five main subcategories. Fatty acids, triglycerides, phospholipids, sphingolipids, and sterol lipids are some of them. The second-highest level of lipids in the human body is found in the brain, which uses all five kinds of lipids extensively [258]. Early brain metabolism declines in AD patients may be caused by a loss of metabolic control and an increase in ROS production [259]. Lipid oxidation is a very initial sign of AD in patients’ brains. Unsaturated lipids are attacked by hydroxy radicals to produce highly reactive secondary products such reactive carbonyls and reactive aldehydes, which can inactivate enzyme active sites and override their physiological function [259]. Also, membranes that have been oxidized have changed mobility. As a result, it is not surprising that antioxidant therapy has been suggested as a cure for AD numerous times.
A well-known antioxidant, singlet oxygen quencher, and lipid metabolism regulator is vitamin E. One of the most significant antioxidants in the brain is vitamin E, particularly the α-tocopherol form. Human carriers of a mutation in the α-TTP gene exhibits progressive spinocerebellar ataxia, areflexia, loss of proprioception, and severely low vitamin E concentrations, underscoring its pivotal role in brain function. Significantly, patients with AD have higher levels of α-TTP expression in their brains [260].
Phosphatidylserine, phosphatidylcholine, phosphatidylethanolamine, as well as phosphatidylinositol make up the cerebral cortex's neuronal membrane phospholipids [261]. Phosphatidylserine is the most abundant acidic phospholipid in the membranes of nerve cells and is particularly crucial for maintaining brain processes [261]. It was noted that this phospholipid, particularly in older people, plays a role in controlling the release of acetylcholine, dopamine, and noradrenaline. The cerebral cortex and retina have the largest concentrations of docosahexaenoic acid [DHA, 22:6 (n-3)], an unsaturated fatty acid present in phospholipids [261]. Variations in phospholipid metabolite concentrations have been linked to cognitive decline and the neuropathological symptoms of AD. Indeed, phospholipids modify the synthesis of A-beta40, A-beta42 peptides and, as a result, A-beta42:40 ratios [262]. A protective eicosanoid called neuroprotectin D1 (NPD1), that has a strong anti-inflammatory and neuroprotective bioactivity, is currently known to be produced by DHA in the brain's phospholipids. NPD1, which inhibits the production of the A-beta42 peptide from aged human brain cells and is substantially reduced in AD patients' brains [262].
The amounts of phospholipids in the cell membranes may alter as a result of vitamin E administration through a variety of possible methods. It has been demonstrated that vitamin E affects gene expression and modifies the activity of several enzymes involved in signal transduction. The enzymes phospholipase A2 and diacylglycerol kinase are among these [263]. Older rats’ hippocampal sphingolipid metabolism was controlled by a- and gamma -tocopherol [263]. Sphingolipids, which are also important elements of the oligodendrocyte membrane, direct the myelination process in the CNS. The regulation of brain sphingolipid metabolism, especially sulfatide metabolism, has also been linked to vitamin K [264]. Age-related cognitive loss is at risk due to myelin sulfatide dysregulation [265]. Sulfatides and vitamin K, which is almost entirely found in the form of MK-4 in the brain, are positively correlated [265].
As the obligatory precursor of CoA, vitamin B5 serves as an essential trace nutrient and is vital for a variety of biological processes. The production of acetylcholine and the intricate fatty-acyl chains of myelin that are necessary for myelin function, particularly the galactosylcerebrosides and galactosylsulphatides that support the insulating function of myelinated neurons, are mediated by CoA in the brain, where they enable saltatory neuronal conduction [266]. In regions with a larger concentration of myelinated structures, cerebral B5 is determined more. This may be because these cells are more active, which would necessitate higher CoA-dependent glucose metabolism to support TCA-cycle activity. The recent research revealed that B5 deficiency may significantly contribute to the cognitive impairment, neurodegeneration, loss of myelin, and lack of acetylcholine that characterizes age-related dementias like AD [267]. In particular, vitamin B12 is assigned a role in the DNA synthesis of oligodendrocytes that produce myelin and the production of myelin [137]. Many nerves’ axons are encased in the myelin sheath, which protects them from electrical current and acts as an electrical insulator to allow for rapid conduction velocity [137]. It greatly aids in the repair of damaged nerves because of its significant role in myelin synthesis and remyelination [137]. Lack of vitamin B12 damages the myelin sheath that surrounds the cranial, spinal, and peripheral nerves [268]. Vitamin B7 may stimulate the production of fatty acids, which would increase re-myelination and energy in neurons and prevent myelin loss and hypoxia-driven neurodegeneration [169].
Choline is an imperative in the biosynthesis of phosphatidylcholine (a major lipid in myelin). It is the direct precursor of sphingomyelin (another major myelin lipid). It is also involved in the homeostatic control of vital signalling elements that directly affect the start of myelination, the compacting of the myelin sheath, and the maintenance of myelin [269]. A postmortem brain sample study that compared the cerebral cortex of AD patients to age-matched controls revealed decreased concentrations of phosphatidylcholine and phosphatidylethanolamine and enhanced concentrations of their metabolites, glycerophosphocholine and glycerophosphoethanolamine, providing proof that phospholipid metabolism is abnormal in AD [270].
5) Neurotransmitter Synthesis and Synaptogenesis
Neurotransmitters are responsible for sending the messages from one neuron to the next. Signals are sent by these neurotransmitters across neuronal synapse and neuromuscular connections [271]. When the proper signal is received, neurotransmitters are typically released into the synapse from synaptic vesicles located beneath the membrane in the axon terminal. These neurotransmitters are endogenously synthesized from amino acids [271]. Acetylcholine is produced from serine, dopamine from L-phenyl alanine/L-tyrosine, glutamate from glutamine, GABA from glutamate by decarboxylation, glutamate from glutamine, serotonin from L-tryptophan, and histaminergic from L-histidine and N-methyl-D-aspartate, from D-aspartic acid and arginine [272]. Except for dopamine, these are the principal neurotransmitters involved in AD pathogenesis. Otto Loewi originally referred to the first neurotransmitter, acetylcholine, as “vagus stuff” because of its capacity to imitate electrical stimulation of the vagus nerve. It is currently understood that it functions as a neurotransmitter in the CNS at several synapses, the neuromuscular junction, and all autonomic ganglia [272]. The most prevalent neurotransmitter in the brain, glutamate, plays a role in learning and memory [272]. AD patients' dying brain cells release excessive glutamate as they decompose. Now, detail discussion is given on ‘how vitamins involve in the synthesis of neurotransmitters.
Enzymes involved in nerve development and the manufacture of neurotransmitters are activated and deactivated by vitamin D in the CSF and the brain. Vitamin D has been shown to influence the manufacture of neurotransmitters and neurotrophic factors, and the CNS has been revealed to have its receptor [273]. Acetylcholine, dopamine, serotonin, and gamma-aminobutyric acid are only a few of the neurotransmitters whose genetic expression is controlled by 1,25(OH)D, particularly in the hippocampus [274]. Acetylcholine is synthesized in two steps i.e., Formation of acetyl coenzyme A from reaction of acetate and CoA (B5), catalyzed by Acetyl CoA synthetase. Then formation of acetylcholine from reaction of choline and Acetyl CoA, catalyzed by choline acetylase called also choline acetyltransferase. The TCA cycle produces a number of significant intermediates. Among them is α-ketoglutarate that may be used to make glutamate. This reaction is brought on by a coenzyme called vitamin B3 (NAD+) and an enzyme called glutamate dehydrogenase [275]. The enzyme glutamate decarboxylase, which uses vitamin B6 as a cofactor, converts precursor glutamate into GABA in the cytoplasm of pre-synaptic neurons. Additionally, the enzyme glutaminase produces glutamate, a neurotransmitter, in the CNS from glutamine as part of the glutamate-glutamine cycle [275]. For the production of monoamines or biogenic neurotransmitters (dopamine, norepinephrine, epinephrine, serotonin, and histamine), two extra cofactors are needed; vitamin B6 is required as a cofactor for decarboxylation that is performed by aromatic amino acid decarboxylase. The storage vesicle needs vitamin C as a complement to convert dopamine to norepinephrine. The monoamine (histamine) is created when the amino acid (histidine) is decarboxylated to produce it all at once [276]. Different enzyme is used to decarboxylate histidine i.e., histidine decarboxylase. This enzyme also requires vitamin B6 [276]. Serotonin (also called as 5-hydroxytryptamine) is synthesized into two steps; in first step, L-tryptophan converted into 5-hydroxytryptophan in the presence of tyrosine hydroxylase; in the second step, Serotonin production from 5-hydroxytryptophan requires vitamin B6 as a cofactor [137].
In the histidine cycle, histidine is deaminated in the presence of B9, producing urocanic acid. This acid participates in a variety of metabolic pathways to produce formiminoglutamate, or also called as"FIGLU," which is then converted into glutamate by the enzyme formiminotransferase [277]. As is well known, the neurotransmitter glutamate is essential for the brain's learning and memory processes. The serine and glycine cycle depends on vitamin B9; 5,10-methylene tetrahydrofolate gives glycine residues a hydroxymethyl group to form serine, which is known to be the main source of the one carbon unit needed in the B9 process [277]. When B9 is lacking, the glycine is unable to make serine, which is necessary for the synthesis of tryptophan, an amino acid that is used to create the neurotransmitter serotonin [137].
6) Amyloidosis and Tau Protein Phosphorylation
Extracellular aggregated A-beta-peptide deposits and intra-neuronal neurofibrillary tangles (NFTs), which are made up of abnormally hyperphosphorylated τ-protein neuronal microtubule-bound protein whose function is controlled by site-specific phosphorylation, are the two main histopathological hallmarks of AD. One of the initial advancements in the progress of AD, occurring before dystrophic neurites and NFTs, is τ-hyperphosphorylation [278]. By boosting the activity of serine/threonine kinases or decreasing the activity of phosphoseryl/phosphothreonyl protein phosphatases, τ can be hyperphosphorylated. Glycogen synthase kinase 3-beta (GSK3-beta) is the primary kinase that phosphorylates-τ in the brain, despite the fact that other protein kinases are known to work on τ [278]. The primary brain serine/threonine protein phosphatases that can dephosphorylate-τ at numerous locations are protein phosphatase 2A (PP2A) [279]. Researchers exposed that a lack of B vitamins, specifically B9 and B12, led to an upregulation of the GSK3-beta and PP2A genes [278]. During this situation, PP2A activity was decreased due to a decrease in the methylated catalytic subunit PP2Ac. In contrast, these scientists noticed an increase in GSK3-beta protein and a change in the phosphorylation sites that control GSK3-beta function [278]. Thus, it appears that the dysregulation of the equilibrium between GSK3-beta and PP2A, which leads in aberrant hyperphosphorylated-τ, is caused by a change in one-carbon metabolism. The effects of ageing in AD patients may be lessened by vitamin D, which has been shown to alleviate age-related τ-hyperphosphorylation and cognitive decline by increasing brain energy metabolism, redox status as well as PP2A activity [280]. It was first discovered in 2000 that scyllo-inositol stabilized the non-toxic oligomers of A-beta and prevented their harmful aggregation in AD subjects [281]. Molecular dynamics simulations of the interaction between scyllo-inositol and simple peptide models were performed to clarify the mechanism by which scyllo-inositol prevents the self-aggregation of A-beta. Scyllo-inositol was found to be able to bind to the surface of A-beta protofibrils in order to stop them from aggregating, but it was unable to dissolve already-formed aggregate [282].
Conclusion
This review indicates to elucidate the function of vitamins in patients of AD. Vitamins have many imperative roles in human physiology. The two most frequently mentioned ways that vitamins can affect cognitive performance in AD are (1) by influences on certain plasma amino acids named Hcy for B vitamins (B6, B9, and B12) and (2) through antioxidant impacts, which lower the levels of free radicals that can harm human cells, for vitamins C, E, D, ß-carotene, and B2. These theories regarding potential processes are mostly focused on preclinical research and findings of links between dementia or cognitive decline and high levels of Hcy or other oxidative stress indicators. Vitamin C prevents the production of nitrosamines through lowering nitrites, vitamin E prevents lipid peroxidation, and carotenes prevent lipid peroxidation. However, a thorough knowledge of processes is needed. Choline, a quasivitamin, works to defend the brain from AD in at least two ways. First, it prevents the development of A-beta plaques, the hallmark pathology of AD. Second, choline supplementation lessens microglia activation, which induces inflammation in the brain and may actually result in nerve cell death, impairing cognitive function. The enzyme beta-secretase-1, which is necessary for converting beta-amyloid precursor protein into amyloid-beta or amyloid-beta oligomerization, can be inhibited by inositols. The Acetyl-L-carnitine has useful effects on dementia and cognition in AD.
The results from current studies show that the effectiveness and protection of vitamin supplementation to avert MCI and the initial stages of AD will likely depend on the following factors: firstly which metabolic pathways are impaired, secondly which vitamins are deficient and could remedy the relevant metabolic flaw, and thirdly, the modulating effect of nutrient-nutrient and nutrient-genotype interaction. To grasp the full potential of vitamin therapy in avoiding dementia and cognitive function as well as to prevent catastrophe, more attention must be paid to a correct dietary strategy.
Author Contributions
The authors have no conflict of interests.
References
- Dey A, Bhattacharya R, Mukherjee A, Pandey DK. Natural products against Alzheimer's disease:Pharmaco-therapeutics and biotechnological interventions. Biotechnology Advances, (2017); 35:178-216.
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Molecular Medicine, (2016); 8(6):595-608.
- Agostinho P, Cunha RA, Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Current Pharmaceutical Design, (2010); 16(25): 2766-2778.
- Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Fore-casting the global burden of Alzheimer’s disease. Alzheimer’s Dementia, (2007); 3:186-191.
- Alzheimer’s disease International. World Alzheimer Report. The Global Economic Impact of Dementia. 2015.
- Hurd MD, Martorell P, Delavande A, Mullen KJ, Langa KM. Monetary costs of dementia in the United States. The New England Journal of Medicine, (2013); 368:1326-1334.
- Di Carlo A, Baldereschi M, Amaducci L, Lepore V, Bracco L, et al. Incidence of dementia, Alzheimer's disease, and vascular dementia in Italy. The Italian Longitudinal Study on Aging (ILSA). Jounal of the American Geriatrics Society, (2002); 50:41-48.
- De-Paula VJ, Radanovic M, Diniz BS, Forlenza OV. Alzheimer's disease. Subcellular Biochemstry, (2012); 65:329-352.
- Pivi1 GAK, Vieira NMdeA, da Ponte, JB, de Moraes DSC, Bertolucci PHF. Nutritional management for Alzheimer’s disease in all stages: mild, moderate, and severe. Nutrire, (2017); 42:1.
- Cauwenberghe CV, Broeckhoven CV, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genetics in Medicine, (2016); 18(5): 421-430.
- Jiang Y, Gao H, Turdu G. Traditional Chinese medicinal herbs as potential AChE inhibitors for anti-Alzheimer’s disease: A review. Bioorganic Chemistry, (2017); 75:50-61.
- Colizzi C. The protective effects of polyphenols on Alzheimer’s disease: A systematic review. Alzheimer’s & Dementia: Translational Research and Clinical Interventions, (2019); 5:184-196.
- Abate G, Marziano M, Rungratanawanich W, Memo M, Uberti D. Nutrition and ageing: focusing on Alzheimer’s disease. Oxidative Medicine and Cell Longivty, (2017); 87(6); 301-317.
- Hu N, Yu J, Tan L, Wang Y, Sun L, et al. Nutrition and the risk of Alzheimer’s disease. BioMed Research International, (2013); 1-12.
- Bhatti AB, Usman M, Ali F, Satti SA. Vitamin supplementation as an adjuvant treatment for Alzheimer’s disease. Journal of Clinical and Diagnostic Research, (2016); 10(8): OE07-OE11.
- Ono K, Yamada M. Vitamin A and Alzheimer’s disease. Geriatrics & Gerontology International, (2012); 12(2):180-188.
- Przybelski RJ, Binkley NC. Is vitamin D important for preserving cognition? A positive correlation of serum 25-hydroxyvitamin D concentration with cognitive function.
- Archives of Biochemistry and Biophysics, (2007); 460(2):202-205.
- Farina N, Isaac MG, Clark AR, Rusted J, Tabet N. Vitamin E for Alzheimer’s dementia and mild cognitive impairment. Cochrane Database of Systematic Review, (2012); 11(11); 78-90.
- Osiezagha K, Ali S, Freeman C, Barker N C, Jabeen S, et al. Thiamine deficiency and delirium. Innovations in Clinical Neuroscience, (2013); 10(4):26-32.
- Pawlak R, Lester SE, Babatunde T. The prevalence of cobalamin deficiency among vegetarians assessed by serum vitamin B12: a review of literature. European Journal of Clinical Nutrition, (2014); 68(5):541-548.
- da Silva SL, Vellas B, Elemans S, Luchsinger J, Kamphuis P, et al. Plasma nutrient status of patients with Al¬zheimer’s disease: systematic review and meta-analysis. Alzheimer’s Demensia, (2014); 10:485-502.
- Lee H-P, Casadesus G, Zhu X, Lee H-g, Perry G, et al. All-trans retinoic acid as a novel therapeutic strategy for Alzheimer’s disease. Expert Review of Neurotherapeutics, (2009); 9:1615-1621.
- Moneim AE. Oxidant/antioxidant imbalance and the risk of Alzheimer’s disease. Current Alzheimer Research, (2015); 12:335-349.
- Green KN, Steffan JS, Martinez-Coria H, Sun X, Schreiber SS, et al. Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. Journal of Neurosciences, (2008); 28:11500-11510.
- Kennedy DO. B vitamins and the brain: mechanisms, dose and efficacy–A review. Nutrients, (2016); 8:68.
- Sánchez-Hernández D, Anderson GH, Poon AN, Pannia E, Cho CE, et al. Maternal fat-soluble vitamins, brain development, and regulation of feeding behavior: an overview of research. Nutrition Research, (2016); 36(10):1045-1054.
- Khillan JS. Vitamin A/retinol and maintenance of pluripotency of stem cells. Nutrients, (2014); 6:1209-1222.
- Hira H, Saleem U, Anwar F, Sohail MF, Raza Z, et al. beta-carotene: A natural compound improves cognitive impairment and oxidative stress in a mouse model of streptozotocin-induced Alzheimer’s disease. Biomolecules, (2019); 9(9):441.
- Ono K, Yamada M. Vitamin A and Alzheimer’s disease. Geriatrics & Gerontology International, 2012; 12(2):180-188.
- Takasaki J, Ono K, Yoshiike Y, Hirohata M, Ikeda T, et al. Vitamin A has antioligomerization effects on amyloid-beta in vitro. Journal of Alzheimer’s disease, (2011); 27(2):271-280.
- Johnson EJ, Vishwanathan R, Johnson MA, Hausman DB, Davey A, et al. Relationship between serum and brain carotenoids, alpha-tocopherol, and retinol concentrations and cognitive performance in the oldest old from the Georgia centenarian study. Journal of Aging Reserch, (2013); 2013:951786. doi: 10.1155/2013/951786
- Feart C, Letenneur L, Helmer C, Samieri C, Schalch W, et al. Plasma carotenoids are inversely associated with dementia risk in an elderly French cohort. Journals of Gerontology: Medical Sciences, (2016); 71(5): 683-688.
- Wang Y, Chung SJ, McCullough ML, Song WO, Fernandez ML, et al. Dietary carotenoids are associated with cardiovascular disease risk biomarkers mediated by serum carotenoid concentrations. Journal of Nutrition, (2014); 144:1067-1074.
- Liu PP, Xie Y, Meng XY, Kang JS. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduction Targeted Therapy, (2019); 4:29.
- Goodman AB, Pardee AB. Evidence for defective retinoid transport and function in late onset Alzheimer’s disease. The Proceedings of the National Academy of Sciences, USA, (2003); 100:2901-2905.
- Kirchmeyer M, Koufany M, Sebillaud S, Netter P, Jouzeau J-Y, et al. All-trans retinoic acid suppresses interleukin-6 expression in interleukin-1-stimulated synovial fibroblasts by inhibition of ERK1/2 pathway independently of RAR activation. Arthritis Research & Therapy, (2008); 10:R141.
- Kaur C, Sivakumar V, Dheen ST, Ling EA. Insulin-like growth factor I and II expression and modulation in amoeboid microglial cells by lipopolysaccharide and retinoic acid. Neurosciences, (2006); 138:1233-1244.
- Behairi N, Belkhelfa M, Rafa H, Labsi M, Deghbar N, e al. All-trans retinoic acid (ATRA) prevents lipopolysaccharide-induced neuroinflammation, amyloidogenesis and memory impairment in aged rats. Journal of Neuroimmunology, (2016); 300:21-29.
- Takamura R, Watamura N, Nikkuni M, Ohshima T. All-trans retinoic acid improved impaired proliferation of neural stem cells and suppressed microglial activation in the hippocampus in an Alzheimer’s mouse model. Journal of Neurosciences Reseach, (2017); 95:897-906.
- Priyanka SH, Syam Das S, Thushara AJ, Rauf AA, Indira M. All trans retinoic acid attenuates markers of neuroinflammation in rat brain by modulation of SIRT1 and NF-κB. Neurochemical Research, (2018); 43:1791-180.
- Das, BC, Dasgupta S, Ray SK. Potential therapeutic roles of retinoids for prevention of neuroinflammation and neurodegeneration in Alzheimer’s disease. Neural Regeneration Research, (2019); 14(11):1880-1892.
- Szutowicz A, Bielarczyk H, Jankowska-Kulawy A, Ronowska A, Pawełczyk T. Retinoic acid as a therapeutic option in Alzheimer’s disease: a focus on cholinergic restoration. Expert Review Neurotherapy, (2015); 15:239-249.
- Kawahara K, Suenobu M, Ohtsuka H, Kuniyasu A, Sugimoto Y, et al. Cooperative therapeutic action of retinoic acid receptor and retinoid x receptor agonists in a mouse model of Alzheimer’s disease. Journal of Alzheimers Disease, (2014); 42:587-605.
- Nourhashemi F, Hooper C, Cantet C, Féart C, Gennero I, et al. Cross-sectional associations of plasma vitamin D with cerebral beta-amyloid in older adults at risk of dementia. Alzheimer's Research Therapy, (2018); 10:43.
- Przybelski RJ, Binkley NC. Is vitamin D important for preserving cognition? A positive correlation of serum 25- hydroxyvitamin D concentration with cognitive function. Archives of Biochemistry & Biophysics, (2007); 460 (2):202-205.
- Sommer I, Griebler U, Kien C, Auer S, Klerings I, et al. Vitamin D deficiency as a risk factor for dementia: a systematic review and meta-analysis. BMC Geriatrics, (2017); 17:16.
- Littlejohns TJ, Kos K, Henley WE, Kuźma E, Llewellyn DJ. Vitamin D and dementia. Journal of Prevention Alzheimer’s Disease, (2016); 3(1):43-52.
- Holick MF. Vitamin D deficiency. New England Journal of Medicine, (2007); 357:266-281.
- Balion C, Griffith LE, Strifler L, Henderson M, Patterson C, et al. Vitamin D, cognition, and dementia: a systematic review and meta-analysis. Neurology, (2012); 79(13):1397-1405.
- Annweiler C, Llewellyn DJ, Beauchet O. Low serum vitamin D concentrations in Alzheimer’s disease: a systematic review and meta-analysis. Journal of Alzheimers Disease, (2013); 33(3):659-74.
- Annweiler C, Annweiler T, Bartha R, Herrmann FR, Camicioli R, et al. Vitamin D and white matter abnormalities in older adults: a cross-sectional neuroimaging study. European Journal of Neurology, (2014); 21(12):1436-e95.
- Shen L, Ji H-F. Vitamin D deficiency is associated with increased risk of Alzheimer’s disease and dementia: evidence from meta-analysis. Nutrition Journal, (2015); 14:76.
- Barnard K, Colon-Emeric C. Extraskeletal effects of vitamin D in older adults: cardiovascular disease, mortality, mood, and cognition. American Journal of Geriatric Pharmacotherapy, (2010); 8(1):4-33.
- Llewellyn DJ, Lang IA, Langa KM, Muniz-Terrera G, Phillips CL, et al. Vitamin D and risk of cognitive decline in elderly persons. Archives of International Medicine, (2010); 170:1135-1141.
- Slinin Y, Paudel ML, Taylor BC, Fink HA, Ishani A, et al. 25-Hydroxyvitamin D levels and cognitive performance and decline in elderly men. Neurology, (2010); 74:33-41.
- Wilson VK, Houston DK, Kilpatrick L, Lovato J, Yaffe K, et al. Relationship between 25-hydroxyvitamin D and cognitive function in older adults: the health, aging and body composition study. Journal of the American Geriatric Society, (2014); 62:636-641.
- Toffanello ED, Coin A, Perissinotto E, Zambon S, Sarti S, et al. Vitamin D deficiency predicts cognitive decline in older men and women: The Progetto Veneto Anziani (Pro.V.A.) Study. Neurology, (2014); 83(24):2292-2298.
- Perna L, Mons U, Kliegel M, Brenner H. Serum 25-hydroxyvitamin D and cognitive decline: a longitudinal study among non-demented older adults. Dementia and Geriatric Cognitive Disorders, (2014); 38:254-263.
- Breitling LP, Perna L, Müller H, Raum E, Kliegel M, et al. Vitamin D and cognitive functioning in the elderly population in Germany. Experimental Gerontology, (2012); 47:122-127.
- Granic A, Hill TR, Kirkwood TBL, Davies K, Collerton J, et al. Serum 25-hydroxyvitamin D and cognitive decline in the very old: the Newcastle 85+ Study. European Journal of Neurology, (2015); 22:106-115.
- Przybelski R, Agrawal S, Krueger D, Engelke JA, Walbrun F, et al. Rapid correction of low vitamin D status in nursing home residents. Osteoporosis International, (2008); 19:1621-1628.
- Stein MS, Scherer SC, Ladd KS, Harrison LC. A randomized controlled trial of high-dose vitamin D2 followed by intranasal insulin in Alzheimer’s disease. Journal of Alzheimer’s Disease, (2011); 26:47-84.
- Annweiler C, Fantino B, Gautier J, Beaudenon M, Thiery S, et al. Cognitive effects of vitamin D supplementation in older outpatients visiting a memory clinic: a pre-post study. Journal of American Geriatric Society, (2012); 60:793-795.
- Rossom RC, Espeland MA, Manson JE, Dysken MW, Johnson KC, et al. Calcium and vitamin D supplementation and cognitive impairment in the women’s health initiative. Journal of American Geriatric Society, (2012); 60:2197-2205.
- Annweiler C, Rolland Y, Schott AM, Blain H, Vellas B, et al. Serum vitamin D deficiency as a predictor of incident non-Alzheimer dementias: a 7-year longitudinal study. Dementia and Geriatric Cognitive Disorders, (2011); 32:273-278.
- Afzal S, Bojesen SE, Nordestgaard BG. Reduced 25-hydroxyvitamin D and risk of Alzheimer’s disease and vascular dementia. Alzheimer’s Dementia, (2014); 10:296-302.
- Littlejohns TJ, Henley WE, Lang IA, Annweiler C, Beauchet O, et al. Vitamin D and the risk of dementia and Alzheimer disease. Neurology, (2014); 83:920-928.
- Knekt P, Sääksjärvi K, Järvinen R, Marniemi J, Männistö S, et al. Serum 25-hydroxyvitamin D concentration and risk of dementia. Epidemiology, (2014); 25:799-804.
- Karakis I, Pase MP, Beiser A, Booth SL, Jacques PF, et al. Association of serum vitamin D with the risk of incident dementia and subclinical indices of brain aging: the framingham heart study. Journal of Alzheimer’s Disease, (2016); 51:451-461.
- Feart C, Helmer C, Merle B, Herrmann FR, Annweiler C, et al. Associations of lower vitamin D concentrations with cognitive decline and long-term risk of dementia and Alzheimer’s disease in older adults. Alzheimer’s Dementia, (2017); 13:1207-1216.
- Licher S, de Bruijn R, Wolters FJ, Zillikens MC, Ikram MA, et al. Vitamin D and the risk of dementia: the Rotterdam study. Journal of Alzheimer’s Disease, (2017); 60:989-997.
- Chen H, Xue W, Li J, Fu K, Shi H, et al. 25-hydroxyvitamin D levels and the risk of dementia and Alzheimer’s disease: A dose–response meta-analysis. Front Aging Neuroscience, (2018); 10:368.
- Olsson E, Byberg L, Karlstrom B, Cederholm T, Melhus H, et al. Vitamin D is not associated with incident dementia or cognitive impairment: an 18-y follow-up study in community-living old men. American Journal of Clinical Nutrition, (2017); 105:936-943.
- Gezen-Ak D, Dursun E, Ertan T, Hanagasi H, Gurvit H, et al. Association between vitamin D receptor gene polymorphism and Alzheimer’s disease. The Tohoku Journal of Experimental Medicine, (2007); 212:275-282.
- Kuningas M, Mooijaart SP, Jolles J, Slagboom PE, Westendorp RG, et al. VDR gene variants associate with cognitive function and depressive symptoms in old age. Neurobiology of Aging, (2009); 30:466-473.
- Lehmann DJ, Refsum H, Warden DR, Medway C, Wilcock GK, et al. The vitamin D receptor gene is associated with Alzheimer’s disease. Neuroscience Letters, (2011); 504: 79-82.
- Laczmanski L, Jakubik M, Bednarek-Tupikowska G, Rymaszewska J, Sloka N, et al. Vitamin D receptor gene polymorphisms in Alzheimer’s disease patients. Experimental Gerontology, (2015); 69:142-147.
- Wang L, Hara K, Van Baaren JM, Price JC, Beecham GW, et al. Vitamin D receptor and Alzheimer’s disease: A genetic and functional study. Neurobiology of Aging, (2012); 33: 1844 e1841-1849.
- Beydoun MA, Ding EL, Beydoun HA, Tanaka T, Ferrucci L, et al. Vitamin D receptor and megalin gene polymorphisms and their associations with longitudinal cognitive change in US adults. American Journal of Clinical Nutrition, (2012); 95:163-178.
- Galli F, Azzi A, Birringer M, Cook-Mills JM, Eggersdorfer M, et al. Vitamin E: Emerging aspects and new directions. Free Radical Biology and Medicine, (2017); 102:16-36.
- Shahidi F, de Camargo AC. Tocopherols and tocotrienols in common and emerging dietary sources: Occurrence, applications, and health benefits. International Journal of Molecular Science, (2016); 17:1745.
- Jiang Q. Natural forms of vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Radical Biology and Medicine, (2014); 72:76-90.
- Ulatowski LM, Manor D. Vitamin E and neurodegeneration. Neurobiology of Disease, (2015); 84: 78-83.
- Ulatowski L, Manor D. Vitamin E trafficking in neurologic health and disease. Annual Review of Nutrition, (2013); 33:87-103.
- Ulatowski L, Dreussi C, Noy N, Barnholtz-Sloan J, Klein E, et al. Expression of the -tocopherol transfer protein gene is regulated by oxidative stress and common single-nucleotide polymorphisms. Radical Biology and Medicine, (2012); 53:2318-2326.
- Reiter E, Jiang Q, Christen S. Anti-inflammatory properties of - and gamma-tocopherol. Molecular Aspects of Medicine, (2007); 28(5-6):668-691.
- Lloret A, Esteve D, Monllor P, Cervera-Ferri A, Lloret A. The Effectiveness of vitamin E treatment in Alzheimer’s disease. International Journal of Molecular Science, (2019); 20: 879.
- Lee CY, Man-FanWan J. Vitamin E supplementation improves cell-mediated immunity and oxidative stress of Asian men and women. Journal of Nutrition, (2000); 130:2932-2937.
- De la Fuente M, Hernanz A, Guayerbas N, Victor VM, Arnalich F. Vitamin E ingestion improves several immune functions in elderly men and women. Free Radical Researsh, (2008); 42:272-280.
- Jiang Z, Yin X, Jiang Q. Natural forms of vitamin E and 130-carboxychromanol, a long-chain vitamin E metabolite, inhibit leukotriene generation from stimulated neutrophils by blocking calcium influx and suppressing 5-lipoxygenase activity, respectively. Journal of Immunology, (2011); 186:1173-1179.
- Lopes da Silva S, Vellas B, Elemans S, Luchsinger J, Kamphuis P, et al. Plasma nutrient status of patients with Alzheimer’s disease: Systematic review and meta-analysis. Alzheimer’s Dementia, (2014); 10:485-502.
- de Wilde MC, Vellas B, Girault E, Yavuz AC, Sijben JW. Lower brain and blood nutrient status in Alzheimer’s disease: Results from meta-analyses. Alzheimer’s Dementia, (2017); 3:416-431.
- Dong Y, Chen X, Liu Y, Shu Y, Chen T, et al. Do low-serum vitamin E levels increase the risk of Alzheimer disease in older people? Evidence from a meta-analysis of case-control studies. International Journal of Geriatric Psychiatry, (2018); 33:e257-e263.
- Liu G, Zhao Y, Jin S, Hu Y, Wang T, et al. Circulating vitamin E levels and Alzheimer’s disease: A Mendelian randomization study. Neurobiology of Aging. (2018); 72:e1-e189.
- Zandi PP, Anthony JC, Khachaturian AS, Stone SV, Gustafson D, et al. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: The Cache County Study. Archives of Neurology, (2004); 61:82-88.
- Devore EE, Grodstein F, van Rooij FJA, Stampfer MJ, Witteman JC, et al. Dietary antioxidants and long-term risk of dementia. Archives of Neurology, (2010); 67(7):819-825.
- Basambombo LL, Carmichael PH, Côté S, Laurin D. Use of vitamin E and C supplements for the prevention of cognitive decline. Annals of Pharmacotherapy, (2017); 51:118-124.
- Gray SL, Anderson ML, Crane PK, Breitner JC, McCormick W, et al. Antioxidant vitamin supplement use and risk of dementia or Alzheimer’s disease in older adults. Journal of American Geriatric Society, (2008); 56:291-295.
- Masaki KH, Losonczy KG, Izmirlian G, Foley DJ, Ross GW, et al. Association of vitamin E and C supplement use with cognitive function and dementia in elderly men. Neurology, (2000); 54:1265-1272.
- Luchsinger JA, Tang MX, Shea S, Mayeux R. Antioxidant vitamin intake and risk of Alzheimer disease. Archives of Neurology, (2003); 60(2):203-208.
- Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. New England Journal of Medicine, (2005); 352:2379-2388.
- Kang JH, Cook N, Manson J, Buring JE, Grodstein F. A randomized trial of vitamin E supplementation and cognitive function in women. Archives of International Medicine, (2006); 166:2462-2468.
- Lloret A, Badía MC, Mora NJ, Pallardó FV, Alonso MD, et al. Vitamin E paradox in Alzheimer’s disease: It does not prevent loss of cognition and may even be detrimental. Journal of Alzheimer’s Disease, (2009); 17:143-149.
- Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA, (2014); 311(1):33-44.
- Kryscio RJ, Abner EL, Caban-Holt A, Lovell M, Goodman P, et al. Association of antioxidant supplement use and dementia in the prevention of Alzheimer’s disease by vitamin E and selenium trial (PREADViSE). JAMA Neurology, (2017); 74:567-573.
- Ibrahima NF, Yanagisawa D, Durani LW, Hamezah HS, Damanhuri HA, et al. Tocotrienol-rich fraction modulates amyloid pathology and improves cognitive function in AbetaPP/PS1 mice. Journal of Alzheimer’s Disease, (2017); 55:597-612.
- Dong S, Huang X, Zhen J, Van Halm-Lutterodt N, Wang J, et al. Dietary vitamin E status dictates oxidative stress outcomes by modulating effects of fish oil supplementation in Alzheimer’s disease model APPswe/PS1dE9 mice. Molecular Neurobiology, (2018); 55:9204-9209.
- Ferland G. Vitamin K and brain function. Seminars in Thrombosis and Hemostasis, (2013); 39:849-855.
- Ferland G, Doucet I, Mainville D. Phylloquinone and menaquinone-4 tissue distribution at different life stages in male and female spraguedawley rats fed different VK levels since weaning or subjected to a 40% calorie restriction since adulthood. Nutrients, (2016); 8:141.
- Ohsaki Y, Shirakawa H, Hiwatashi K, Furukawa Y, Mizutani T, et al. Vitamin K suppresses lipopolysaccharide-induced inflammation in the rat. Bioscience, Biotechnology and Biochemistry, (2006); 70:926-632.
- Ohsaki Y, Shirakawa H, Miura A, Giriwono PE, Sato S, et al. Vitamin K suppresses the lipopolysaccharide-induced expression of inflammatory cytokines in cultured macrophage-like cells via the inhibition of the activation of nuclear factor κB through the repression of IKKalpha/beta phosphorylation. Journal of Nutrition and Biochemistry, (2010); 21:1120-1126.
- Carrié I, Bélanger E, Portoukalian J, Rochford J, Ferland G. Lifelong lowphylloquinone intake is associated with cognitive impairments in old rats. Journal of Nutrition, (2011); 141:1495-1501.
- Sato Y, Honda Y, Hayashida N, Iwamoto J, Kanoko T, et al. Vitamin K deficiency and osteopenia in elderly women with Alzheimer’s disease. The Archives of Physical Medicine and Rehabilitation, (2005); 86:576-581.
- Presse N, Belleville S, Gaudreau P, Greenwood CE, Kergoat MJ, et al. Vitamin K status and cognitive function in healthy older adults. Neurobiology of Aging, (2013); 34(12): 2777-2783.
- van den Heuvel EGHM, van Schoor NM, Vermeer CRML, Zwijsen MH, Comijs HC. Vitamin K status is not associated with cognitive decline in middle aged adults. Journal of Nutrition Health and Aging, (2015); 19:908-912.
- Chouet J, Ferland G, Féart C, Rolland Y, Presse N, et al. Dietary vitamin K intake is associated with cognition and behaviour among geriatric patients: the CLIP study. Nutrients, (2015); 7:6739-6750.
- Soutif-Veillon A, Ferland G, Rolland Y, Presse N, Boucher K, et al. Increased dietary vitamin K intake is associated with less severe subjective memory complaint among older adults. Maturitas, (2016); 93:131-136.
- Kiely A, Ferland G, Ouliass B, O’Toole PW, Purtill H, et al. Vitamin K status and inflammation are associated with cognition in older Irish adults. Nutritional Neuroscience, (2020); 23(8):591-599.
- Van Gorp RH, Schurgers LJ. New insights into the pros and cons of the clinical use of vitamin K antagonists (VKAs) versus direct oral anticoagulants (DOACs). Nutrients, (2015); 7:9538-9557.
- Alisi L, Cao R, De Angelis C, Cafolla A, Caramia F, et al. The relationships between vitamin K and cognition: A review of current evidence. Frontiers in Neurology, (2019); 10:239.
- Ferland G, Doucet I, Mainville D. Phylloquinone and menaquinone-4 tissue distribution at different life stages in male and female spraguedawley rats fed different VK levels since weaning or subjected to a 40% calorie restriction since adulthood. Nutrients, (2016); 8:141.
- Brangier A, Celle S, Roche F, Beauchet O, Ferland G, et al. Use of vitamin K antagonists and brain morphological changes in older adults: an exposed/unexposed voxel-based morphometric study. Dementia and Geriatric Cognitive Disorders, (2018); 45:18-26.
- Cafolla A, Gentili A, Cafolla C, Perez V, Baldacci E, et al. Plasma vitamin K1 levels in Italian patients receiving oral anticoagulant therapy for mechanical heart prosthesis: a case-control study. American Journal of Cardiovascuular Drugs, (2016); 16:267-274.
- Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther MG, et al. Pharmacology and management of the vitamin K antagonists:American college of chest physicians’ evidence-based clinical practice guidelines (8th edition). Chest, (2008); 133(6):160S-198S.
- Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, et al. Alzheimer’s disease. Lancet, (2016); 388:505-517.
- Morris MC, Schneider JA, Tangney CC. Thoughts on B-vitamins and dementia. Journal of Alzheimer’s Disease, (2006); 9(4):429-433.
- Smith AD, Smith SM, de Jager CA,Whitbread P, Johnston C, et al. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: a randomized controlled trial. PLoS One, (2010); 5:e12244.
- Oulhaj A, Jernerén F, Refsum H, Smith AD, de Jager CA. Omega-3 fatty acid status enhances the prevention of cognitive decline by B vitamins in mild cognitive impairment. Journal of Alzheimer’s Disease, (2016); 50:547-557.
- Khosravi-Largani M, Pourvali-Talatappeha P, Roustaa AM, Karimi-Kivia M, Noroozia E, et al. A review on potential roles of vitamins in incidence, progression, and improvement of Multiple Sclerosis. eNeurologicalScience, (2018); 10:37-44.
- Vauzour D, Camprubi-Robles M, Miquel-Kergoat S, Andres-Lacueva C, Bánáti D, et al. Nutrition for the ageing brain: Towards evidence for an optimal diet. Ageing Research Reviews, (2017); 35:222-240.
- Obeid R, Herrmann W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Letters, (2006); 580:2994-3005.
- O’Leary F, Allman-Farinelli M, Samman S. Vitamin B12 status, cognitive decline and dementia: a systematic review of prospective cohort studies. British Journal of Nutrition, (2012); 108:1948-1961.
- Ford AH, Almeida OP. Effect of homocysteine lowering treatment on cognitive function: a systematic review and meta-analysis of randomized controlled trials. Journal of Alzheimer’s Disease, (2012); 29:133-149.
- Malouf R, Areosa Sastre A. Vitamin B12 for cognition. Cochrane Database Systematic Reviews, (2003); 3:CD004326.
- Malouf R, Grimley Evans J. Folic acid with or without vitamin B12 for the prevention and treatment of healthy elderly and demented people. Cochrane Database Systematic Reviews, (2008); 4: CD004514.
- Durga J, van Boxtel MP, Schouten EG, Kok FJ, Jolles J, et al. Effect of 3-year folic acid supplementation on cognitive function in older adults in the FACIT trial: a randomized, double blind, controlled trial. Lancet, (2007); 369(9557):208-216.
- Calderón-Ospina CA, Nava-Mesa MO. B vitamins in the nervous system: Current knowledge of the biochemical modes of action and synergies of thiamine, pyridoxine, and cobalamin. CNS Neuroscience Therapy, (2020); 26:5-13.
- da Silva SL, Vellas B, Elemans S, Luchsinger J, Kamphuis P, et al. Plasma nutrient status of patients with Al¬zheimer’s disease:systematic review and meta-analysis. Alzheimer’s Dementia, (2014); 10:485-502.
- Ulstein I, Bøhmer T. Normal vitamin levels and nutritional indices in Alzheimer’s disease patients with mild cognitive impairment or dementia with normal body mass indexes. Journal of Alzheimer’s Disease, (2017); 55:717-725.
- Froese DS, Fowler B, Baumgartner MR. Vitamin B12, folate and the methionine remethylation cycle-biochemistry, pathways and regulation. Journal of Inherited Metabolic Disese, (2019); 42:673-685.
- Green R, Allen LH, Bjørke-Monsen A-L, Brito A, Gueant J-L, et al. Vitamin B12 deficiency. Nature Reviews Disease Primers, (2017); 3:17040.
- Moore EM, Ames D, Mander AG, Carne RP, Brodaty H, et al. Among vitamin B12 deficient older people, high folate levels are associated with worse cognitive function: combined data from three cohorts. Journal of Alzheimer’s Disease, (2014); 39:661-668.
- Grarup N, Sulem P, Sandholt CH, Thorleifsson G, Ahluwalia TS, et al. Genetic architecture of vitamin B12 and folate levels uncovered applying deeply sequenced large datasets. Plos Genetics, (2013); 9:e1003530.
- Larsson SC, Traylor M, Malik R, Dichgans M, Burgess S, et al. Modifiable pathways in Alzheimer’s disease: Mendelian randomization analysis. BMJ, (2017); 359:j5375.
- Taliun SAG. Genetic determinants of low vitamin B12 levels in Alzheimer’s disease risk. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring, (2019); 11: 430-434.
- Ikram MA, Vrooman HA, Vernooij MW, den Heijer T, Hofman A, et al. Brain tissue volumes in relation to cognitive function and risk of dementia. Neurobiology of Aging, (2010); 31:378-386.
- Gallucci M, Spagnolo P, Arico M, Grossi E. Predictors of response to cholinesterase inhibitors treatment of Alzheimer’s disease: date mining from the TREDEM registry. Journal of Alzheimer’s Disease, (2016); 50:969-979.
- Gibson GE, Hirsch JA, Fonzetti P, Jordan BD, Cirio RT, et al. Vitamin B1 (thiamine) and dementia. Annals of the New York Academy of Sciences, (2016); 1367:21-30.
- Pan X, Gong N, Zhao J, Yu Z, Gu F, et al. Powerful beneficial effects of benfotiamine on cognitive impairment and beta-amyloid deposition in amyloid precursor protein/presenilin-1 transgenic mice. Brain, (2010); 133:1342-1351.
- Pan X, Fei G, Lu J, Jin L, Pan S, et al. Measurement of blood thiamine metabolites for Alzheimer’s disease diagnosis. EBioMedicine, (2016); 3:155-162.
- Pan X, Sang S, Fei G, Jin L, Liu H, et al. Enhanced activities of blood thiamine diphosphatase and monophosphatase in Alzheimer’s disease. PLoS One, (2017); 12: e0167273.
- Liu, D., Zunji, K. and Jia, L. Thiamine deficiency and neurodegeneration: the interplay among oxidative stress, endoplasmic reticulum stress, and autophagy. Pharmacology and Nutritional Sciences Faculty Publications, (2017); 91.
- Saedisomeolia A, Ashoori M. Riboflavin in human health: a review of current evidences. Advances in Food and Nutrition Research, (2018); 83:57-81.
- Kim H, Kim G, Jang W, Kim SY, Chang N. Association between intake of B vitamins and cognitive function in elderly Koreans with cognitive impairment. Nutrition Journal, (2014); 13:118.
- Araki A, Yoshimura Y, Sakurai T, Umegaki H, Kamada C, et al. Low intakes of carotene, vitamin B2, pantothenate and calcium predict cognitive decline among elderly patients with diabetes mellitus: the Japanese elderly diabetes intervention trial. Geriatric Gerontology International, (2017); 17:1168-1175.
- Tao L, Liu K, Chen S, Yu H, An Y, et al. Dietary intake of riboflavin and unsaturated fatty acid can improve the multi-domain cognitive function in middle-aged and elderly populations: A 2-year prospective cohort study. Frontiers in Aging Neuroscience, (2019); 11:226.
- Zhao R, Wang H, Qiao C, Zhao K. Vitamin B2 blocks development of Alzheimer’s disease in APP/PS1 transgenic mice via anti-oxidative mechanism. Tropical Jounal of Pharmacological Research, (2018); 17(6):1049-1054.
- Cui X, Chopp M, Zacharek A, Roberts C, Buller B, e al. Niacin treatment of stroke increases synaptic plasticity and axon growth in rats. Stroke, (2010); 41:2044-2049.
- Morris MC, Evans DA, Bienias JL, Scherr PA, Tangney CC, et al. Dietary niacin and the risk of incident Alzheimer’s disease and of cognitive decline. Journal of Neurology and Neurosurgery Psychiatry, (2004); 75:1093-1099.
- Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, et al. Mitophagy and Alzheimer’s disease: Cellular and molecular mechanisms. Trends in Neurosciences, (2017); 40:151-166.
- Wang X, Hu X, Yang Y, Takata T, Sakurai T. Nicotinamide mononucleotide protects against beta-amyloid oligomer-induced cognitive impairment and neuronal death. Brain Research, (2016); 1643:1-9.
- Kim EJ, Yang SJ. Nicotinamide reduces amyloid precursor protein and presenilin 1 in brain tissues of amyloid beta-tail vein injected mice. Clinical Nutrition Research, (2017); 6:130-135.
- Jesko H, Wencel P, Strosznajder RP, Strosznajder JB. Sirtuins and their roles in brain aging and neurodegenerative disorders. Neurochemical Research, (2017); 42:876-890.
- Rizzi L, Roriz-Cruz M. Sirtuin 1 and Alzheimer’s disease: An up-to-date review. Neuropeptides, (2018); 71:54-60.
- Leonardi R, Zhang YM, Rock CO, Jackowski S. Coenzyme A. Back in action. Progress in Lipid Research, (2005); 44(2e3):125e53.
- Lee J-H, Ahn S-Y, Lee HA, Won KS, Chang HW, et al. Dietary intake of pantothenic acid is associated with cerebral amyloid burden in patients with cognitive impairment. Food and Nutrition Research, (2018); 62:1415.
- Sato K. Why is vitamin B6 effective in alleviating the symptoms of autism? Medical Hypotheses, (2018);115:103-106.
- Zhuo J-M, Wang H, Praticò D. Is hyperhomocysteinemia an Alzheimer’s disease (AD) risk factor, an AD marker or neither? Trends in Pharmacological Sciences, (2011); 32(9): 562-571.
- Moretti R, Caruso P. The Controversial role of homocysteine in neurology: From labs to clinical practice. International Journal of Molecular Science, (2019); 20(1):231.
- Tinelli C, Pino AD, Ficulle E, Marcelli S, Feligioni M. Hyperhomocysteinemia as a risk factor and potential nutraceutical target for certain pathologies. Frontiers in Nutrition, (2019); 6:49.
- Miller JW, Green R, Mungas DM, Reed BR, Jagus WJ. Homocysteine, vitamin B6, and vascular disease in AD patients. Neurology, (2002); 58(10).
- Tong L. Structure and function of biotin-dependent carboxylases. Cellular and Molecular Life Sciences, (2013); 70(5):863-891.
- McCarty MF, DiNicolantonio JJ. Neuroprotective potential of high-dose biotin. Medical Hypotheses, (2017); 109:145-149.
- Palmeri A, Ricciarelli R, Gulisano W, Rivera D, Rebosio C, et al. Amyloid-beta peptide is needed for cGMP-induced long-term potentiation and memory. Journal of Neurosciences, (2017); 37(29):6926-6937.
- Gonos ES, Kapetanou M, Sereikaite J, Bartosz G, Naparło K, et al. Origin and pathophysiology of protein carbonylation, nitration and chlorination in age-related brain diseases and aging. Aging, (2018); 10:868-901.
- Kang WB, Chen YJ, Lu DY, Yan JZ. Folic acid contributes to peripheral nerve injury repair by promoting Schwann cell proliferation, migration, and secretion of nerve growth factor. Neural Regeneration Research, (2019); 14(1):132-139.
- Chen H, Liu S, Ji L, Wu T, Ji Y, et al. Folic Acid supplementation mitigates Alzheimer’s disease by reducing inflammation:A randomized controlled trial. Mediators of Inflammation, (2016); 14(3): 135-144.
- Morris MC, Evans DA, Bienias JL, Tangney CC, Hebert LE, et al. Dietary folate and vitamin B12 intake and cognitive decline among community-dwelling older persons. Archives of Neurology, (2005); 62(4):641-645.
- Ma F, Wu T, Zhao J, Song A, Liu H, et al. Folic acid supplementation improves cognitive function by reducing the levels of peripheral inflammatory cytokines in elderly Chinese subjects with MCI. Scientific Reports, (2016); 6:37487.
- Hankey GJ, Ford AH, Yi Q, Eikelboom JW, Lees KR, et al. VITATOPS trial study group. Effect of B vitamins and lowering homocysteine on cognitive impairment in patients with previous stroke or transient ischemic attack: a prespecified secondary analysis of a randomized, placebo-controlled trial and meta-analysis. Stroke, (2013); 44: 2232-2239.
- Li MM, Yu JT, Wang HF, Jiang T, Wang J, et al. Efficacy of vitamins B supplementation on mild cognitive impairment and Alzheimer's disease: a systematic review and meta-analysis. Current Alzheimer Research, (2014); 11(9):844-852.
- Love S, Miners JS. Cerebrovascular disease in ageing and Alzheimer’s disease. Acta Neuropathology, (2016); 131:645-658.
- Kruman II, Kumaravel TS, Lohani A, Pedersen WA, Cutler RG, et al. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. Journal of Neuroscience, (2002); 22:1752-1762.
- Corradaa MM, Kawasa CH, Hallfrischc J, Mullerd D, Brookmeyer R. Reduced risk of Alzheimer’s disease with high folate intake: The Baltimore longitudinal study of aging. Alzheimers Dementia, (2005); 1(1):11-18.
- Almeida CC, Brentani HP, Forlenza OV, Diniz BS. Serum folic acid is reduced in patients with Alzheimer’s disease. Revista de Psiquiatria Clínica, (2012); 39(3):90-93.
- Harrison FE, Bowman GL, Polidori MC. Ascorbic acid and the brain: rationale for the use against cognitive decline. Nutrients, (2014); 6:1752-1781.
- Monacelli F, Acquarone E, Giannotti C, Borghi R, Nencioni A. Vitamin C, aging and Alzheimer’s disease. Nutrients, (2017); 9:670.
- Murakami K., Murata N, Ozawa Y, Kinoshita N, Irie K, et al. Vitamin C restores behavioral deficits and amyloid-beta-oligomerization without affecting plaque formation in a mouse model of Alzheimer’s disease. Journal of Alzheimer’s Disease, (2011); 26(1):7-18.
- Luchsinger JA, Tang M-X, Shea S, Mayeux R. Antioxidant vitamin intake and risk of Alzheimer’s disease. Archives of Neurology, (2003); 60(2):203-208.
- Zandi PP, Anthony JC, Khachaturian AS, Stone SV, Gustafson D, et al. Reduced risk of Alzheimer’s disease in users of antioxidant vitamin supplements: The cache county study. Archives of Neurology, (2004); 61:82-88.
- Harrison FE. A critical review of vitamin C for the prevention of age-related cognitive decline and Alzheimer’s disease. Journal of Alzheimer’s Disase, (2012); 29(4):711-726.
- Choudhry F, Howlett DR, Richardson JC, Francis PT, Williams RJ. Pro-oxidant diet enhances beta/gamma secretase-mediated APP processing in APP/PS1 transgenic mice. Neurobiology of Aging, (2012); 33:960-968.
- Covarrubias-Pinto A, Acuna AI, Beltran FA, Torres-Diaz L, Castro MA. Old things new view: Ascorbic acid protects the brain in neurodegenerative disorders. International Journal of Molecular Science, (2015); 16:28194-28217.
- Rohl C, Armbrust E, Herbst E, Jess A, Gulden M, et al. Mechanisms involved in the modulation of astroglial resistance to oxidative stress induced by activated microglia: Antioxidative systems, peroxide elimination, radical generation, lipid peroxidation. Neurotoxicity Research, (2010); 17:317-331.
- Lam V, Hackett M, Takechi R. Antioxidants and dementia risk: Consideration through a cerebrovascular perspective. Nutrients, (2016); 8(12):828.
- Quinn J, Suh J, Moore MM, Kaye J, Frei B. Antioxidants in Alzheimer’s disease-vitamin C delivery to a demanding brain. Journal of Alzheimer’s Disease, (2003); 5:309-313.
- Devore EE, Kang JH, Stampfer MJ, Grodstein F. The association of antioxidants and cognition in the nurses’ health study. American Journal of Epidemiology, (2013); 177: 33-41.
- Fillenbaum GG, Kuchibhatla MN, Hanlon JT, Artz MB, Pieper CF, et al. Dementia and Alzheimer’s disease in community-dwelling elders taking vitamin C and/or vitamin E. Annals of Pharmacotherapy, (2005); 39:2009-2014.
- Emir UE, Raatz S, McPherson S, Hodges JS, Torkelson C, et al. Noninvasive quantification of ascorbate and glutathione concentration in the elderly human brain. NMR Biomedicine, (2011); 24:888-894.
- González HF, Visentin S. Micronutrients and neurodevelopment: An update. Archivos Argentinos de Pediatria, (2016); 114(6):570-575.
- Tayebati SK, Amenta F. Choline-containing phospholipids: relevance to brain functional pathways. Clinical Chemistry and Laboratory Medicine, (2013); 51:513-521.
- Shea, T.B. Choline and phosphatidylcholine may maintain cognitive performance by multiple mechanisms. American Journal of Clinical Nutrition, (2019); 110(6):1268-1269.
- Pohanka, M. Alpha7 nicotinic acetylcholine receptor is a target in pharmacology and toxicology. International Journal of Molecular Science, (2012); 13(2):2219-2238.
- Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, et al. The role of microglia in the healthy brain. Journal of Neuroscience, (2011); 31(45):16064-16069.
- Jin JL, Fang M, Zhao YX, Liu XY. Roles of sigma-1 receptors in Alzheimer's disease. Int J Clin Exp Med. 2015; 8(4):4808-4820. PMCID: PMC4484039
- Matsumura A, Suzuki S, Iwahara N, Hisahara S, Kawamata J, et al. Temporal changes of CD68 and alpha7 nicotinic acetylcholine receptor expression in microglia in Alzheimer’s disease-like mouse models. Journal of Alzheimer's Disease, (2015); 44(2):409-423.
- Brailoiu E, Chakraborty S, Brailoiu GC, Zhao P, Barr JL, et al. Choline is an intracellular messenger linking extracellular stimuli to IP3-evoked Ca(2+) signals through sigma-1 receptors. Cell Reports, (2019); 26(2):330-337 e4.
- Hall AA, Herrera Y, Ajmo CTJr, Cuevas J, Pennypacker KR. Sigma receptors suppress multiple aspects of microglial activation. Glia, (2009); 57(7):744-754.
- US Standing Committee on the Scientific Evaluation. US standing committee on the scientific evaluation of dietary reference intakes and its panel on folate, other B vitamins, and choline dietary reference intakes for thiamin, riboflavin, niacin, vitamin B6, folate, vitamin B12, pantothenic acid, biotin, and choline, (1998); Washingdon, DC:The National Academies Collection.
- Velazquez R, Ferreira E, Knowles S, Fux C, Rodin A, et al. Lifelong choline supplementation ameliorates Alzheimer’s disease pathology and associated cognitive deficits by attenuating microglia activation. Aging Cell, (2019); 18:e13037.
- Caudill MA, Strupp BJ, Muscalu L, Nevins JEH, Canfield RL. Maternal choline supplementation during the third trimester of pregnancy improves infant information processing speed: A randomized, double-blind, controlled feeding study. FASEB Journal, (2018); 32(4):2172-2180.
- Secades JJ. Citicoline in the treatment of cognitive impairment. Journal of Neurology and Experimental Neuroscience, (2019); 5(1):14-26.
- McGlade E, Agoston AM, DiMuzio J, Kizaki M, Nakazaki E, et al. The effect of citicoline supplementation on motor speed and attention in adolescent males. Journal of Attention Disorders, (2019); 23(2):121-134.
- Bruce SE, Werner KB, Preston BF, Baker LM. Improvements in concentration, working memory and sustained attention following consumption of a natural citicoline-caffeine beverage. International Journal of Food Science and Nutrition, (2014); 65:1003-1007.
- De Jesus MMM. Cognitive improvement in mild to moderate Alzheimer’s dementia after treatment with the acetylcholine precursor choline alfoscerate: A multicenter, double-blind, randomized, placebo-controlled trial. Clinical Therapy, (2003); 25(1):178-193.
- Amenta F, Parnetti L, Gallai V, Wallin A. Treatment of cognitive dysfunction associated with Alzheimer’s disease with cholinergic precursors. Ineffective treatments or inappropriate approaches? Mechanisms of Ageing and Development, (2001); 122(16):2025-2040.
- Scapicchio PL. Revisiting choline alphoscerate profile: a new, perspective, role in dementia? International Journal of Neuroscience, (2013); 123(7):444-449.
- Higgins JP, Flicker L. Lecithin for dementia and cognitive impairment. Cochrane Database of Systematic Reviews, (2003); (3):CD001015.
- Chheri, DR. Myo-Inositol and Its Derivatives: Their emerging role in the treatment of human diseases. Frontiers in Pharmacology, (2019); 10:1172.
- Siger M, Schuff N, Zhu X, Miller BL, Weiner MW. Regional myo-inositol concentration in mild cognitive impairment using 1H magnetic resonance spectroscopic imaging. Alzheimer Disease and Associated Disorders, (2009); 23(1):57-62.
- Lee D, Lee W-S, Lim S, Kim YK, Jung H-Y, Das S, Lee J, Luo W, Kim K-T, Chung S-K. A guanidine-appended scylloinositol derivative AAD-66 enhances brain delivery and ameliorates Alzheimer’s phenotypes. Scientific Repotrts, (2017); 7(1):14125.
- Fenili D, Weng Y-Q , Aubert I, Nitz M , McLaurin J. Sodium/myo-Inositol Transporters: Substrate Transport Requirements and Regional Brain Expression in the TgCRND8 Mouse Model of Amyloid Pathology. PLoS ONE, (2011); 6(8):e24032.
- Lyketsos C, Abushakra S, Liang E, Hernandez R, Tariot P, et al. Effects of oral ELND005 (Scylloinositol) on neuropsychiatric symptoms in a 78-week phase 2 study in mild/ moderate Alzheimer’s disease (AD): potential role of Myo-inositol reduction. Alzheimer's & Dementia: Journal of the Alzheimer's Association, (2012); 8(4S):S771-S772.
- Liang E, Tariot P, Abushakra S, Porsteinsson A, Hernandez R, et al. Effects of oral ELND005 (Scylloinositol) on brain Scyllo-inositol and Myo-inositol levels: spectroscopy results from a phase 2 study in mild-tomoderate Alzheimer’s disease. Alzheimer's & Dementia: Journal of the Alzheimer's Association, (2012); 8(4S):S783-S784.
- Ribas GS, Vargas CR, Wajner M. L-carnitine supplementation as a potential antioxidant therapy for inherited neurometabolic disorders. Gene, (2014); 533:469-76.
- Flanagan JL, Simmons PA, Vehige J, Willcox MDP, Garrett Q. Review role of carnitine in disease. Nutrition & Metabolism (Lond), (2010); 7:30.
- Liu J, Head E, Gharib AM, Yuan W, Ingersoll RT, et al. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha -lipoic acid. Proceedings of the National Academy of Sciences of the USA, (2002); 99:2356-2361.
- Pettegrew JW, Klunk WE, Panchalingam K, Kanfer JN, McClure RJ. Clinical and neurochemical effects of acetyl-L-carnitine in Alzheimer's disease. Neurobiology of Aging, (1995); 16:1-4.
- Hudson S, Tabet N. Acetyl-L-carnitine for dementia. Cochrane Database of Systematic Reviws, (2003); (2):CD003158.
- Cristofano A, Sapere N, Marca GL, Angiolillo A, Vitale M, et al. Serum levels of acyl-carnitines along the continuum from normal to Alzheimer's dementia. PLoS One, (2016); 11(5):e0155694.
- Onofrj M, Ciccocioppo F, Varanese S, di Muzio A, Calvani M, et al. Acetyl-L-carnitine: from a biological curiosity to a drug for the peripheral nervous system and beyond. Expert Review of Neurotherapy, (2013); 13:925-936.
- Thal LJ, Calvani M, Amato A, Carta A. A 1-year controlled trial of acetyl-l-carnitine in early-onset AD. Neurology, (2000); 55:805-810.
- Montgomery SA, Thal LJ, Amrein R. Meta-analysis of double blind randomized controlled clinical trials of acetyl-L-carnitine versus placebo in the treatment of mild cognitive impairment and mild Alzheimer's disease. International Clinical Psychopharmacology, (2003); 18:61-71.
- Yang YS, Choi H, Lee CN, Kim YB, Kwak YT. A multicenter, randomized, double blind, placebo-controlled clinical trial for efficacy of acetyl-L-carnitine in patients with dementia associated with cerebrovascular disease. Dementia and Neurocognitive Disorders, (2018); 17(1):1-10.
- Fenech M. Vitamins associated with brain aging, mild cognitive impairment, and Alzheimer disease: biomarkers, epidemiological and experimental evidence, plausible mechanisms, and knowledge gaps. Advances in Nutrition, (2017); 8:958-970.
- Kennedy DO. B vitamins and the brain: mechanisms, dose and efficacy– a review. Nutrients, (2016); 8:68.
- Araújo JR, Martel F, Borges N, Araújo JM, Keating E. Folates and aging: Role in mild cognitive impairment, dementia and depression. Ageing Research Reviews, (2015); 22: 9-19.
- Fuso A, Seminara L, Cavallaro RA, D'Anselmi F, Scarpa S. S-adenosylmethionine / homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Molecular and Cellular Neuroscience, (2005); 28(1):195-204.
- Porter K, Hoey L, Hughes CF, Ward M, McNulty H. Causes, consequences and public health implications of low B-vitamin status in ageing. Nutrients, (2016); 8(11):725.
- Serot J-M, Barbe´ F, Arning E, Bottiglieri T, Franck P, et al. Homocysteine and methylmalonic acid concentrations in cerebrospinal fluid: relation with age and Alzheimer’s disease. Joural of Neurology and Neurosurgey Psychiatry, 2005; 76:1585-1587.
- Herrmann W, Schorr H, Bodis M, Knapp JP, Müller A, et al. Role of homocysteine, cystathionine and methylmalonic acid measurement for diagnosis of vitamin deficiency in high-aged subjects. European Journal of Clinical Investigation, (2000); 30:1083-1089.
- Maynard S, Fang EF, Scheibye-Knudsen M, Croteau DL, Bohr VA. DNA damage, DNA repair, aging, and neurodegeneration. Cold Spring Harbor Perspectives in Medicine, (2015); 5:a025130.
- Madabhushi R, Pan L, Tsai L-H. DNA damage and its links to neurodegeneration. Neuron, (2014); 83(2):266-282.
- Gruz-Gibelli E, Chessel N, Allioux C, Marin P, Piotton F, et al. The vitamin A derivative all-trans retinoic acid repairs amyloid-beta-induced double-strand breaks in neural cells and in the murine neocortex. Neural Plasticity, (2016); Article ID 3707406, 11 pages.
- Kim GH, Kim JE, Rhie SJ Yoon S. The Role of oxidative stress in neurodegenerative diseases. Experimental Neurobiology, (2015); 24(4):325-340.
- Reddy PV, Perry G, Cooke MS, Sayre LM, Smith MA. Mechanisms of DNA Damage and Repair in Alzheimer Disease. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience, (2000-2013).
- Sajjad N, Wani A, Hassan S, Ali R, Hamid R, et al. Interplay of antioxidants in Alzheimer’s disease. Jounal of Translational Science, (2019); 5:1-11.
- Wimalawansa SJ. Vitamin D deficiency: Effects on oxidative stress, epigenetics, gene regulation, and aging. Biology (Basel), (2019); 8:30.
- Surjana D, Halliday GM, Damian DL. Role of nicotinamide in DNA damage, mutagenesis, and DNA repair. Journal of Nucleic Acids, (2010); 20(10): 190-204.
- Ladeira C, Carolino E, Gomes MC, Brito M. Role of macronutrients and micronutrients in DNA damage:Results from a food frequency questionnaire. Nutrition and Metabolic Insights, (2017); 1-8.
- Karapiperi K, Gousis C, Papaioannidou P. The role of vitamin B12 in DNA modulation mechanisms. Front. Pharmacol. Conference Abstract: 8th Southeast European Congress on Xenobiotic Metabolism and Toxicity-XEMET (2010); 18-20.
- Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer's disease. Oxid Med Cell Longevity, (2013); 2013:316523.
- Wang X, Wang W, Li L, Perry G, Lee HG, et al. Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochimica et Biophysica Acta (2014); 1842(8):1240-1247.
- Berridge MJ. Vitamin D deficiency accelerates ageing and age-related diseases: a novel hypothesis. Journal of Physiology, (2017); 595(22):6825-6836.
- Scaini G, Rezin GT, Carvalho AF, Streck EL, Berk M, et al. Mitochondrial dysfunction in bipolar disorder: Evidence, pathophysiology and translational implications. Neuroscience and Biobehavioral Reviews, (2016); 68:694-713.
- Depeint F, Bruce WR, Shangari N, Mehta R, O’Brien PJ. Mitochondrial function and toxicity: Role of the B vitamin family on mitochondrial energy metabolism. Chemico-Biological Interactions, (2006); 163:94-112.
- Janssen JJE, Grefte S, Keijer J, de Boer VCJ. Mito-Nuclear Communication by Mitochondrial Metabolites and Its Regulation by B-Vitamins. Frontiers in Physiology, (2019); 10:78.
- Hamilton JA, Hillard CJ, Spector AA, Watkins PA. Brain uptake and utilization of fatty acids, lipids and lipoproteins: application to neurological disorders. Journal of Molecular Neuroscience, (2007); 33:2-11.
- Mosconi L, Murray J, Davies M, Williams S, Pirraglia E, et al. Nutrient intake and brain biomarkers of Alzheimer’s disease in at-risk cognitively normal individuals: a cross-sectional neuroimaging pilot study. BMJ Open, (2014); 4:e004850.
- Lloret A, Esteve D, Monllor P, Cervera-Ferri A, Lloret A. The Effectiveness of vitamin E treatment in Alzheimer’s disease. International Journal of Molecular Science, (2019); 20:879.
- Langelier B, Linard A, Bordat C, Lavialle M, Heberden C. Long chain-polyunsaturated fatty acids modulate membrane phospholipids composition and protein localization in lipid rafts of neural stem cell cultures. Journal of Cellular Biochemistry, (2010); 15: 1356-1364.
- Schaefer EJ, Bongard V, Beiser AS, Lamon-Fava S, Robins SJ, et al. Plasma phosphatidylcholine docosahexaenoic acid content and risk of dementia and Alzheimer’s disease: the Framingham Heart Study. Archives of Neurology, (2006); 63(5); 147-155.
- Hassouneh LKM. Effects of vitamin E on the synthesis of phospholipids and brain functions in old rats. Neurophysiology, (2018); 50(3):166-172.
- Shearer MJ, Newman P. Recent trends in the metabolism and cell biology of vitamin K with special reference to vitamin K cycling and MK-4 biosynthesis. Journal of Lipid Research, (2014); 55:345-362.
- Crivello NA, Casseus SL, Peterson JW, Smith DE, Booth SL. Age- and brain-region-specific effects of dietary vitamin K on myelin sulfatides. Journal of Nutrition and Biochemistry, (2010); 21(11):1083-1088.
- Aggarwal S, Yurlova L, Simons M. Central nervous system myelin: structure, synthesis and assembly. Tends in Cell Biology, (2011); 21(10):585-592.
- Ismail N, Kureishy N, Church SJ, Scholefield M, Unwin RD, et al. Vitamin B5 (D-pantothenic acid) localizes in myelinated structures of the rat brain: Potential role for cerebral vitamin B5 stores in local myelin homeostasis. Biochemical and Biophysical Research Communications, 2020; 522(1):220-225.
- Briani C, Dalla Torre C, Citton V, Manara R, Pompanin S, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients, (2013); 5(11):4521-39.
- Heffernan C, Jain MR, Liu T, Kim H, Barretto K, et al. Nectin-like 4 complexes with choline transporter-like protein-1 and regulates schwann cell choline homeostasis and lipid biogenesis in vitro. Journal of Biological Chemisty, 2017; 292(11):4484-4498.
- Blusztajn JK, Slack BE, Mellott TJ. Neuroprotective actions of dietary choline. Nutrients, (2017); 9(8):815.
- McEnery MW, Siegel RE. Encyclopedia of the neurological sciences. 2. Academic press; Oxford. Neurotransmitter receptors, (2014):552-564.
- Kandimalla R, Reddy PH. Therapeutics of neurotransmitters in Alzheimer’s disease. Journal of Alzheimers Disease, (2017); 57(4):1049-1069.
- Anjum I, Jaffery SS, Fayyaz M, Samoo Z, Anjum S. The Role of vitamin D in brain health: A mini literature review. Cureus, (2018); 10(7):e2960.
- Banerjee A, Khemka VK, Ganguly A, Roy D, Ganguly U, et al. Vitamin D and Alzheimer’s disease: Neurocognition to therapeutics. International Journal of Alzheimer’s Disease, (2015); 20(1): 89-100.
- Plaitakis A, Kalef-Ezra E, Kotzamani D, Zaganas I, Spanaki C. The glutamate dehydrogenase pathway and its roles in cell and tissue biology in health and disease. Biology, (2017); 6(11): 45-59.
- Best J, Nijhout HF, Samaranayake S, Hashemi P, Reed M. A mathematical model for histamine synthesis, release, and control in varicosities. Theoretical Biology and Medical Modelling, (2017); 14:24.
- Mahmood L. The metabolic processes of folic acid and vitamin B12 deficiency. Journal of Health and Research Reviews, (2014); 1(1):5-9.
- Nicolia V, Fuso A, Cavallaro RA, Luzio AD, Scarpa S. B vitamin deficiency promotes tau phosphorylation through regulation of GSK3beta and PP2A. Journal of Alzheimer’s Disease, (2010); 19:895-907.
- Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, et al. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease. Journal of Biological Chemistry, (2000); 275:5535-5544.
- Briones TL, Darwish H. Decrease in age-related tau hyperphosphorylation and cognitive improvement following vitamin D supplementation are associated with modulation of brain energy metabolism and redox state. Neuroscience, (2014); 262:143-155.
- McLaurin J, Golomb R, Jurewicz A, Antel JP, Fraser PE. Inositol stereoisomers stabilize an oligomeric aggregate of Alzheimer amyloid beta peptide and inhibit a beta-induced toxicity. Journal of Biological Chemistry, (2000); 275(24):18495-18502.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0