

Full Length Research Article
Coding Genome Sequence and Protein Sequence Analysis of Dengue Strains: In Silico Correlation
Aasia Zahid1, Ayesha Afzaal1, Hina Awais*1, Talha Mannan2, Huma Habib1
Adv. life sci., vol. 10, no. 1, pp. 48-53, March 2023
*– Corresponding Author: Hina Awais (Email: hina.awais@mlt.uol.edu.pk)
Authors' Affiliations
2. Department of Medical Lab Technology, Shalamar School of Allied Health Sciences, Lahore – Pakistan
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: DENV-1, DENV-2, DENV-3, and DENV-4 are the four serotypes of dengue viruses (DENV) that are transferred from person to person through the bite of Aedes mosquitoes. Dengue fever has surged 30-fold in occurrence over the last 50 years, making it one of the world's most serious arboviral diseases. The aim of this study was to bioinformatically correlate the coding sequences of four DENV strains to check their genetic & functional diversity based on the similarity of the sequences.
Methods: The coding sequences (CDs) and protein sequences of newly reported dengue strains (DENV 1, DENV 2, DENV 3, and DENV 4) were obtained from the National center for Biotechnology Information (NCBI) nucleotide and protein databases. We compared the genetic and functional compatibility of selected gene sequences from four dengue strains by using various bioinformatics tools and software such as BLAST, MEGA 11.0, ProtParam, GOR4 and SWISS Model.
Results: The total number of amino acids in dengue strains DENV-1, 2, 3, and 4 is 3392, 3391, 3390, and 3387, according to Physicochemical analysis. The phylogenetic analysis reveals that DENV-1 and DENV-2 have more genetic similarity than DENV-2 and DENV-3, with bootstrap values greater than 90%. While different percentages of alpha helices were predicted in secondary structure, such as 33.23%, 36.51%, 31.21%, and 32.27% of DENV-1, 2, 3, and 4 show little variation. The non-structural proteins NS1 and NS5 of all four DENV strains show more than 65 percent similarity index in 3D structure analysis.
Conclusion: This study first presented a bioinformatics comparison of all four DENV strains. The 3D and 2D structures of DENV strains (1-4) show some similarity and dissimilarity index, however the four DENV strains differ in their 2D structure's alpha helix (H), random coil, and number of amino acids.
Keywords: Bioinformatics; Dengue strains; Non-structural protein; Coding sequence; viral pathogenesis
Introduction![]()
The dengue virus (DENV) causes dengue fever (DF), the most common arthropod-borne viral disease in humans. Both dengue fever and dengue hemorrhagic fever are severe viral illnesses that have the possibility of spreading widely. In 1994, there was the first reported dengue epidemic in Karachi. With one death, a total of 145 people were admitted to the hospital. Haripur district saw the onset of illness in 2003. It resulted in mortality for 700 people. The DENV 2 serotype was responsible for this outbreak [1]. According to a study published by the WHO, the dengue virus infects more than 50 million individuals worldwide each year. Dengue hemorrhagic fever (DHF) is diagnosed in 1% of these individuals, and in this population, the disease results in mortality in 4% of incidents [2].
DENV belongs to the Flaviviridae family of viruses, which includes those, that cause yellow fever and the Japanese, West Nile encephalitides and St. Louis [3]. The four serotypes of dengue viruses are DENV-1, DENV-2, DENV-3, and DENV-4, which are transmitted to humans through Aedes mosquito bites, particularly Aedes aegypti and Aedes albopictus[4]. According to the World Health Organization (WHO), up to 100 million illnesses occur each year. Dengue fever has surged 30-fold in occurrence over the last 50 years, making it one of the world’s most serious arboviral diseases [5].
Dengue fever includes three stages: the febrile period (two–seven days), the critical or leaking phase (24–48 hours), and the convalescence phase (2–7 days).The febrile phase is the start of a high fever produced by dengue viremia that lasts 2–7 days. Around the time of chronic phase, which lasts for 24–48 hours, signs of plasma leak appear, such as pleural effusion, ascites, petechiae, or cerebral bleeding [6].
The disease can show in a variety of ways, from asymptomatic or moderate infection to dengue hemorrhagic fever (DHF) with variable degrees of thrombocytopenia and vascular leakage, severe shock syndrome, and multi-organ failure[7].
Dengue fever (DF), dengue shock syndrome (DSS) and dengue hemorrhagic fever (DHF) are all caused by the dengue virus. The resulting viral particles are circular in shape and include a single-stranded, positive-sense RNA molecule with a 5-methyl cap and a single open reading frame, measuring 11 kb in length and 40-50 nm in diameter [8]. Two untranslated sequences (5′ and 3′ UTRs) surround the open reading frame, which are needed for efficient translation and are evolutionarily conserved across flaviviruses. Because the RNA-dependent RNA polymerase encoded by NS5 lacks proofreading, replication of viral genomes is subject to error, resulting in a tremendous variety of genes and antigens between DENV (1-4) Strains [9]. Despite the fact that the four dengue viruses are genetically identical (they share about 65-70 amino acid sequence similarity), there is considerable genetic variation within a single serotype [10]. DENV genome variation, sub-genomic RNA, antibody-dependent enhancement (ADE), memory cross-reactive T cells, anti-DENV NS1 antibodies, and autoimmunity have all been linked to dengue pathogenesis [11]. After an infection with the same serotype, a person is susceptible to re-infection with such a serotype. Because heterologous immunity is only temporary, it’s possible to get infected with a different serotype later [12,13]. So, for that purpose we design this study to know about the similarity and dissimilarity index in the sequences of dengue strains.
Methods![]()
Various bioinformatics tools and software were used to conduct genomic and proteomic analyses on chosen gene sequences from dengue strains.
Collection of Sequences:
The coding sequences (CDs) and protein sequences of dengue strains (DENV 1, DENV 2, DENV 3, and DENV 4 serotypes) were obtained from nucleotide database and protein database, available in NCBI (https://www.ncbi.nlm.nih.gov/). We collected CDs sequences of newly reported four dengue strains 1, 2, 3 and 4 with accession numbers (NC_001477.1, MT982148.1, MZ544588.1, and KU513441.1 respectively). Similarly, the full polyprotein amino acid sequences of all strains were retrieved from the protein database with accession numbers NP_059433.1 (DENV-1), QNQ18280.1 (DENV-2), QXM02604.1 (DENV-3), and ANK35834.1 (DENV-4). For specific protein modeling, NS1 sequences were separately retrieved with accession numbers AHK09922.1 (DENV-1), AAD11533.1 (DENV-2), YP_001531169.1 (DENV-3), and AUF73704.1 (DENV-4) [14].
BLAST (Basic Local Alignment Search Tool):
The nucleotide and amino acid sequences of each serotype (1, 2, 3, and 4) strain were compared to recently published sequences using BLASTn and BLASTp (NCBI).
Multiple sequence alignment:
Both nucleotide and protein sequences were saved in FASTA format. MEGA 11 (Molecular Evolutionary Genetics Analysis Version 11.0) [15] was used for the alignment of nucleotide and amino acid sequences separately, by ClustalW alignment method [16].
Phylogenetic analysis:
Two phylogenetic trees were constructed of DENV-1 and 2 and DENV-2 and 3 to evaluate the genetic and functional compatibility between these strains by using neighbor joining method [17] in MEGA 11.0 software (https://www.megasoftware.net/) [18].
Open reading frame (ORF):
ORF finder NCBI [19] (https:/www.ncbi.nlm.nih.gov/orffinder) was used for the determination of open reading frame of protein sequences among all four strains of dengue (DENV-1, DENV2, DENV3 and DENV4)
Physicochemical analysis of protein:
ProtParam tool was used to calculate the total number of amino acids, molecular weight, instability index, aliphatic index, theoretical isoelectric point(PI), extinction coefficient and GRAVY (Grand average of hydropathicity) of four dengue strains (https://web.expasy.org/protparam/) [20].
Protein structure prediction:
GOR4 online tool was used to assess the secondary structural elements in RNA based sequences of DENV 1, 2, 3 and 4 (https://npsa-prabi.ibcp.fr/NPSA/npsa_gor4.html) [21]. While to construct tertiary structure of dengue virus, SWISS Model was applied. (https://swissmodel.expasy.org/) [22].
Results![]()
Retrieval of nucleotide and protein sequences of dengue strains:
The nucleotide and protein sequences of all four dengue serotypes were collected from NCBI. Total ten sequences, five for nucleotide and five for amino acid of DENV-1 serotype were collected from NCBI. Similarly, same number of each sequence of every serotype (DENV-2, DENV-3 and DENV-4) were retrieved. The sequences were selected based on greater identity with respect to their families through BLAST. Both nucleotide and protein sequences were saved in FASTA format. For the construction of phylogenetic trees, multiple sequence alignment is necessary. ClustalW alignment method was used to align the sequences of both nucleotide and protein.
Phylogenetic Analysis
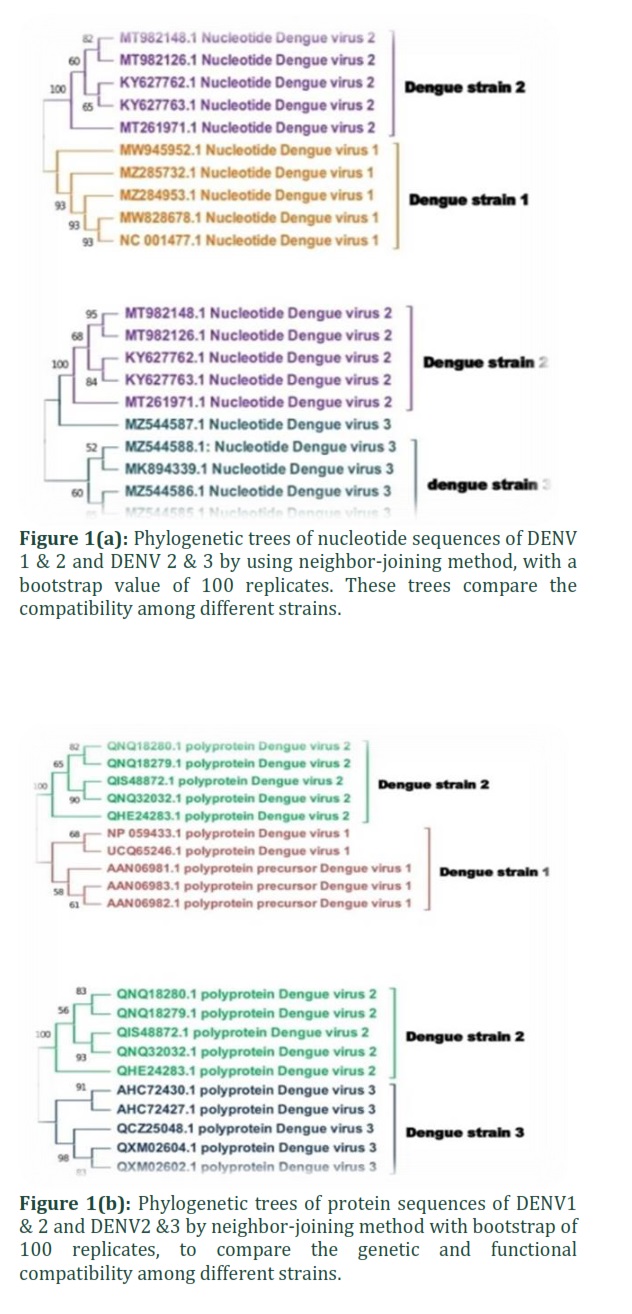
To compare the genetic and functional compatibility among different strainsDENV-1 and DENV-2 and DENV2 and DENV3, the neighbor-joining method was used to create phylogenetic trees, with a bootstrap value of 100 replicates in MEGA11 software (figure 1 (a,b)). The analysis involved total five nucleotide and protein sequences of each serotype. All the nucleotide sequences of DENV-1, 2 and 3 fell into the dengue family. Similarly, all amino acid sequences taken from these strains belong to the same clade. The overall mean distance of nucleotide and protein sequence of DENV-1, 2 was 15.80 and 0.58 while DENV-2, 3 has 10.20 and 1.05 respectively. Average nucleotide similarity correlated with the distance of ≤ 0.5 [23] King and his coworkers reported a study that the largest P-distance of nucleotide variation among DENV-1, DENV-3 and DENV4strains is 5.84%. In contrast, the largest p-distance of amino acid diversity among the full-length genomes is 3.13% [24].
Physicochemical Analysis of protein:
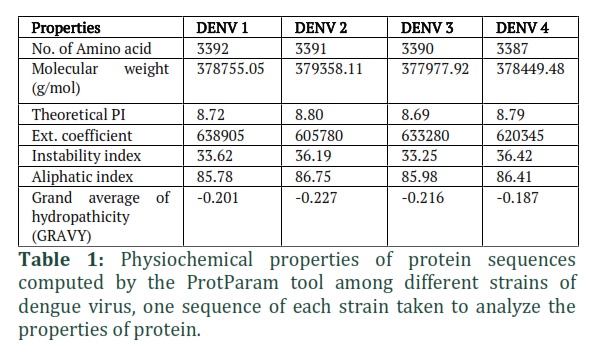
To compute physicochemical properties of protein, ProtParam tool was used. Different parameters of protein sequences were evaluated in this analysis, mentioned in table 1. While ORFfinder NCBI(https:/www.ncbi.nlm.nih.gov/orffinder)was used to determine the open reading frame (ORF) of the protein sequences [25]. In DENV-1 sequence, length of (ORF1) is based on 10179 nucleotides and 3392 amino acids; similarly, 10176nt/3391aa (ORF7) in DENV2, 10173nt/3390aa (ORF1) in DENV3 and 10164nt/3387aa (ORF1) in DENV4 was determined.
One sequence of each strain was taken to analyze the properties of protein. Total number of amino acids in dengue strains i.e., DENV-1, 2, 3 and 4 are 3392, 3391, 3390 and 3387 respectively with isoelectric points (theoretical pI) 8.72, 8.80, 8.69 and 8.79 accordingly. Similarly, their molecular weights range from 377977.92 to 379358.11. Extinction coefficient (605780 to 638905) indicates the amount of light absorbed by a protein at a specific wavelength. The stability of protein estimated by instability index, which is 33.25 to 36.42, these all proteins of DENV strains are stable because their values are less than 40. The aliphatic index revealed that our specified protein was thermo-stable, as the aliphatic side chain had a beneficial effect on globular proteins thermostability. (AI) of these proteins were 85.78 (DENV-1), 86.75 (DENV-2), 85.98(DENV-3) and 86.41 (DENV-4). Protein solubility is indicated by the GRAVY index. The negative values of all four strains (-0.201, -0.227, -0.216, -0.187 respectively) indicated that these proteins are hydrophilic in nature.
Prediction of secondary structure:
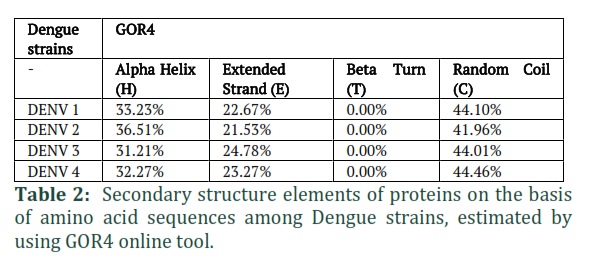
Secondary structures of proteins were estimated based on amino acid sequences by using GOR4 online tool. In the selected protein sequences, the structural elements such as alpha helix, extended strand, beta turn and random coil were determined by this tool as shown in table 2. In results, 0.00% beta sheets of all strains were computed. While different percentages of alpha helices were predicted such as 33.23%, 36.51%, 31.21% and 32.27% of DENV-1, 2, 3 and 4 respectively. Extended strands ranged from 21.53% to 24.78% and coils were estimated in range of 41.96% to 44.96%, showing little diversity.
3D Structure Prediction of protein from amino acid sequences
Sequence collection to predict 3D structure
NS1 non-structural protein sequences in FASTA format of DENV-1 with accession number (AHK09922) , DENV-2 (AAD11533) , DENV-3 (YP_001531169) , DENV-4 (AUF73704) and NS5 proteins sequences with accession number DENV-1 (NP_059433), DENV-2 (QNQ18280.1), DENV-3 (P27915.1), DENV-4 (ANK35834.1) collected from NCBI were used as a template to construct a 3D models for Dengue viruses NS1 and NS5 protein.
Homology modeling using Swiss Model
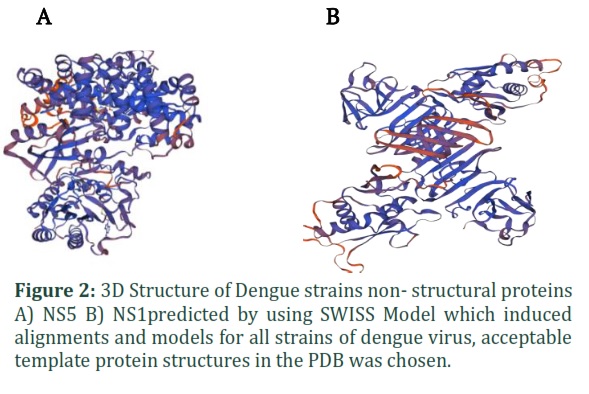
Homology modelling is a method for predicting a protein's 3D structure from its amino acid sequence using a software program. The SWISS-MODEL software was used to induce alignments and homology models for all the strains of dengue virus. First, we choose acceptable template protein structures in the PDB, keeping in mind the following criteria: high coverage (> 65 percent of target aligned to template) and high sequence identity (> 30 percent). Then, as an initial criterion, we employed the Global Model Quality Estimate (GMQE) and Qualitative Model Energy analysis (QMEAN4) scoring functions to distinguish between good and bad models. During modelling, appropriate alignment values were obtained, as well as higher GMQE and QMEAN4 scores, indicating that statistically acceptable homology models were constructed for 2 proteins NS1 and NS5. As all the DENV strains shows 65% of similarity index so the 3D model for each strain is almost same. Figure 2(A, B) shows the 3D structure of NS1 and NS5 (non-structural proteins).
Figures & Tables
The dengue virus (DENV) causes dengue fever (DF), the most common viral disease in humans. The four serotypes of dengue viruses (DENV-1, DENV-2, DENV-3, and DENV-4) are transferred from person to person through the bite of Aedes mosquitoes. In the present study, for analyzing molecular and genomic data of all strains, nucleotide coding sequences and identical protein sequences were retrieved from nucleotide database and protein database respectively, available in NCBI. As in 2020, Dang and his colleagues obtained whole genome sequence of seven DENV2 isolates to analyze genetic diversity among them [26]. Baranoti and his colleagues in 2016 also reported the complete coding sequence of two DENV2 strains that codes polyprotein translated into structural and non-structural proteins [27]. The sequences of all serotypes were selected after comparing with already reported sequences by using BLAST [28].
In the current study, first time four different strains of dengue discussed by using bioinformatics tools to determine the genetic relationships and to compare the genetic and functional compatibility among different strains (DENV-1 and DENV-2) and (DENV-2 and DENV-3),phylogenetic trees were constructed by using neighbor-joining method in MEGA11 software. Similarly, in previously reported study,Huang and his colleagues used MEGA to perform phylogenetic analysis among dengue strains but they construct the tree by using maximum likelihood method [29]. We computed physicochemical properties of protein such as molecular weights and number of amino acids by using Protparam Expasy tool. Aliphatic index, instability index and GRAVY index values determined the stability of proteins, as in 2018, Awais and his colleagues used ExPASy ProtParam for the evaluation of different parameters of HMG CoA synthase like protein sequence [30]. Ali et al. also reported a study in which this tool was used for physicochemical analysis to investigate dengue genome. They estimated the value of aliphatic index more than 70 which indicates the protein thermostability, and negative value of GRAVY index which shows that protein is hydrophilic in nature [31]. Similarly, in our study, the results also concluded the high thermostability and hydrophilic nature of protein in dengue strains as the hydrophilic nature of protein plays a significant role in coil to helix transition and helix-helix assembly which leads to stability.
Currently, the secondary structure of protein was predicted through GOR4 tool. Alpha helix, beta sheet, extended strands and coils were found in structure. In 2017, Ali and his fellows also used online server PSIPRED to estimate helix and beta strands of 457 amino acids sequence of protein derived from dengue genome [31]. We could not use this tool because of large number of amino acids in protein sequence. In this study, prediction of beta sheets was 0.00%, while alpha helix, coils and strands exhibit little diversity among all dengue strains. In 2021, Garrepelly et al. also used GOR4 tool to predict secondary structure elements in plant species and revealed that beta turns of species shows 0.00% amount [32].
To construct 3D structure of protein SWISS Model (Homology modeling server) was used. In the present study, tertiary structure of both NS1 and NS5 proteins among different dengue strains were predicted by keeping the criteria, more than 65% coverage and 30% sequence identity. GMQE and QMEAN4 scores were employed to construct statistically acceptable homology model. As in 2015, Paul and his colleagues reported a study in which they also predict tertiary structure of non-structural protein of dengue using SWISS-PDB viewer, by keeping QMEAN score as an output of models [32].
All the four dengue serotypes were compared bioinformatically to check the genetic diversity and functional compatibility. Many other parameters and tools can be used for further molecular characterization and evolutionary relationships among these strains.
This study presented a bioinformatics comparison of all four DENV strains on the basis of nonstructural protein. The similarity index in 3D structure of NS1 and NS5 of all four strains is greater than 65%, and the beta sheet in secondary structure of all four strains is nearly identical. However, the four DENV strains differ in their 2D structure's alpha helix (H), random coil, and number of amino acids. Current evidence is insufficient to conclude that these differences are responsible for differences in the pathogenicity, immunity, and severity of disease caused by the four DENV strains. Further research will be conducted on the motifs and domain of the DENV genome sequences to determine that which parameter is responsible for the difference in the pathogenicity of four DENV strains.
Author Contributions
Aasia Zahid: Sequence analysis, Write-up. Ayesha Afzaal: Sequence analysis,Write-up. Hina Awais: Conceptualization, Review, Proof read. Talha Mannan: Conceptualization, Review, Proof read. Huma Habib: Writ-up.
![]()
The authors declare that there is no conflict of interest.
![]()
References
- Durrani M, Aslamkhan M, Babar Alam M, Akhtar M, Hanif A. Transmission and epidemic of dengue and its abatement in islamabad capital territory. Pakistan. Journal of Innovative Sciences, (2016); 2(2):21-36.
- Naeem S, Pari A, Gulzar N, Yousaf S, Akhtar MS. Mortality rate of patients with dengue hemorrhagic fever. Pakistan Journal of Medical & Health Sciences, (2018); 12(1):337-339.
- Ross TM. Dengue virus. Clinics in Laboratory Medicine,(2010); 30(1):149-160.
- Long SS, Prober CG, Fischer M. Chapter 2. Principles and practice of pediatric infectious diseases e-book. 2022 of Publication year; Elsevier Health Sciences.
- Cheng NM, Sy CL, Chen BC, Huang TS, Lee SSJ, et al. Isolation of dengue virus from the upper respiratory tract of four patients with dengue fever. PLoS Neglected Tropical Diseases, (2017); 11(4): 1-10.
- Kalayanarooj S. Clinical manifestations and management of dengue/dhf/dss. Tropical Medicine and Health, (2011); 39(4): 83-87.
- Rajapakse S. Dengue shock. Journal of Emergencies, Trauma and Shock, (2011); 4(1):120-127.
- Islam MT, Quispe C, Herrera BJ, Sarkar C, Sharma R, et al. Production, transmission, pathogenesis, and control of dengue virus: A literature-based undivided perspective. BioMed Research International,(2021); 10(4): 1-23.
- Tamura T, Zhang J, Madan V, Biswas A, Schwoerer MP, et al. Generation and characterization of genetically and antigenically diverse infectious clones of dengue virus serotypes 1-4. Emerging Microbes & Infections, (2022); 11(1): 227-239.
- Guzman MG, Halstead SB, Artsob H, Buchy P, Farrar J, et al. Dengue: A continuing global threat. Nature Reviews Microbiology, (2010); 8(12):7-16.
- Haider A, Ullah F, Bilal M, Saif Z, Awais H, et al. Clinical correlation of dengue strains on the basis of seroprevalence in a tertiary care hospital. Pakistan BioMedical Journal, (2022); 5 (4): 149-153.
- Bhatt P, Sabeena SP, Varma M, Arunkumar G. Current understanding of the pathogenesis of dengue virus infection. Current Microbiology, (2021); 78(1):17-32.
- Sasmono RT, Kalalo LP, Trismiasih S, Denis D, Yohan B, et al. Hayati RF, Haryanto S. Multiple introductions of dengue virus strains contribute to dengue outbreaks in east kalimantan, Indonesia, in 2015–2016. Virology Journal, (2019); 16(1):1-15.
- Sayers EW, Beck J, Bolton EE, Bourexis D, Brister JR, et al. Database resources of the national center for biotechnology information. Nucleic Acids Research, (2021); 49(1):10-17.
- Tamura K, Dudley J, Nei M, Kumar S. Mega4: Molecular evolutionary genetics analysis (mega) software version 4.0. Molecular biology and evolution, (2007); 24(8):1596-1599.
- Thompson JD, Higgins DG, Gibson TJ. Clustal W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic acids research, (1994); 22(22):4673-4680.
- Saitou N, Nei M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Molecular biology and evolution, (1987); 4(4):406-425.
- Newman L, Duffus ALJ, Lee C. Using the free program mega to build phylogenetic trees from molecular data. The American Biology Teacher, (2016); 78(7):608-612.
- Rombel IT, Sykes KF, Rayner S, Johnston SA. Orf-finder: A vector for high-throughput gene identification. Gene, (2002); 282(1-2):33-41.
- Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, et al. Expasy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Research, (2003); 31(13):3784-3788.
- Kouza M, Faraggi E, Kolinski A, Kloczkowski A. The GOR method of protein secondary structure prediction and its application as a protein aggregation prediction tool. Prediction of protein secondary structure, (2017); 148(4): 7-24.
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, et al. Swiss-model: Homology modelling of protein structures and complexes. Nucleic Acids Research, (2018); 46(1):296-303.
- Dang TT, Pham MH, Bui HV, Van Le D. Whole genome sequencing and genetic variations in several dengue virus type 1 strains from unusual dengue epidemic of 2017 in Vietnam. Virology journal, (2020); 17(1):1-10.
- King CC, Chao DY, Chien LJ, Chang GJJ, Lin TH, et al. Comparative analysis of full genomic sequences among different genotypes of dengue virus type 3. Virology journal, (2018); 5(63):1-13.
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, et al. Database resources of the national center for biotechnology information. Nucleic Acids Research, (2005); 33(1):39-45.
- Dang TT, Pham MH, Bui HV, Van Le D. First full-length genome sequence of dengue virus serotype 2 circulating in Vietnam in 2017. Infection and Drug Resistance, (2020); 13 (10):4061-4068.
- Baronti C, Piorkowski G, Touret F, Charrel R, Lamballerie X, et al. Complete coding sequences of two dengue virus type 2 strains isolated from an outbreak in Burkina Faso in 2016. Genome Announcements, (2017); 5(17):209-217.
- Huang JH, Su CL, Yang CF, Liao TL, Hsu TC, et al. Molecular characterization and phylogenetic analysis of dengue viruses imported into Taiwan during 2008–2010. The American Journal of Tropical Medicine and Hygiene, 2012; 87(2):349.
- Awais H, Mushtaq Z, Sarwar S, Jamil A. 2018. Sequence analysis of HMG-CoA synthase like gene from Brassica rapa. Cloning & Transgenesis, (2018); 7 (1): 1-7.
- Ali M, Pandey RK, Khatoon N, Narula A, Mishra A, et al. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing Immuno-informatics approach to battle against dengue infection. Scientific Reports, (2017); 7(1):1-13.
- Garrepelly JP, Kiranmayi P, Bandari T, Erva RR. Molecular approach towards screening of biological targets of berberine and its production sources. Current Trends in Biotechnology and Pharmacy, (2021); 15(2):141-152.
- Paul A, Vibhuti A, Raj S, Shaw J. Homology modelling of ns4b protein of dengue using SWISS-PDB viewer. Indian Journal of Pharmaceutical Science & Research, (2015); 5 (4):205-211.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0