

Short Communication
First Step with R for Life Sciences: Learning Basics of this Tool for NGS Data Analysis
Rashid Saif1,2,3*, Kinza Qazi4, Saeeda Zia3,6, Tania Mahmood2, Aniqa Ejaz2, Talha Tamseel4, Suliman Mohammad Alghanem5, Adnan Khaliq7
Adv. life sci., vol. 6, no. 4, pp. 187-194, August 2019
*– Corresponding Author: Rashid Saif (Email: rashid.saif37@gmail.com)
Authors' Affiliations
2. Institute of Biotechnology, Gulab Devi Educational Complex, Lahore, Pakistan
3. Decode Genomics, 264-Q, Johar Town, Lahore, Pakistan
4. Department of Computational Biology and Bioinformatics, Virtual University of Pakistan, Pakistan
5. Department of Biology, University of Tabuk, Tabuk, Kingdom of Saudi Arabia
6. Department of Sciences and Humanities, National University of Computer and Emerging Sciences, Lahore, Pakistan
7. Department of Food Science & Technology, Khwaja Fareed University of Engineering and Information Technology, Rahim Yar Khan, Pakistan
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: R is one of the renowned programming language which is an open source software developed by the scientific community to compute, analyze and visualize big data of any field including biomedical research for bioinformatics applications.
Methods: Here, we outlined R allied packages and affiliated bioinformatics infrastructures e.g. Bioconductor and CRAN. Moreover, basic concepts of factor, vector, data matrix and whole transcriptome RNA-Seq data was analyzed and discussed. Particularly, differential expression workflow on simulated prostate cancer RNA-Seq data was performed through experimental design, data normalization, hypothesis testing and downstream investigations using EdgeR package. A few genes with ectopic expression were retrieved and knowhow to gene enrichment pathway analysis is highlighted using available online tools.
Results: Data matrix of (4×3) was constructed, and a complex data matrix of Golub et al., was analyzed through χ2 statistics by generating a frequency table of 15 true positive, 4 false positive, 15 true negative and 4 false negative on gene expression cut-off values, and a test statistics value of 10.52 with 1 df and p= 0.001 was obtained, which reject the null hypothesis and supported the alternative hypothesis of “predicted state of a person by gene expression cut-off values is dependent on the disease state of patient” in our data. Similarly, sequence data of human Zyxin gene was selected and a null hypothesis of equal frequencies was rejected.
Conclusion: Machine-learning approaches using R statistical package is a supportive tool which can provide systematic prediction of putative causes, present state, future consequences and possible remedies of any problem of modern biology.
Keywords: NGS data; R language; Zyxin gene
Introduction![]()
Statistical analysis has anchored its roots in the field of modern biology and inevitable approach in exploring, manipulating, testing and inferring biological data. R statistical package has opened new avenues due to its flexibility of built-in functions and compatibility with bioconductor packages. Moreover, it helps us to analyze data and providing suitable hypothesis testing in diagnostic and research paraphernalia. Data sets, which were previously difficult to handle and draw conclusions are now easily manipulated and analyzed. R allows univariate data analysis and helps to avoid long and hefty algorithmic approaches. It provides visual manifestations of the data through various graphs and data presentation tools, such as boxplots, heat maps, histograms, quantile-quantile plot (Q-Q Plot), RPKMS plots (Read Per Kilo-base Per Million Mapped Reads), MDS plots (multidimensional scaling), hierarchical clustering, coverage and Manhattan plots. Besides, R is also an important tool in hypothesis testing such as z-test, t-test, χ2-test, Wilcoxon rank test and many more. Thousands of R statistical packages are available for scientific and research data analyses. Recent packages of R with their eminent features are, Infer (An R package that together with the tidyverse framework, works for statistical inference), janitor -simple package in R that examines and cleans data brought by other packages, Esquisse -RStudio add-in that explores data using plots (bar charts, histogram, scatter plots etc.) with ggplot2 package, DataExplorer -Abets in automatic data analysis/predictive modelling, Sparklyr -An exceptional R package that connects to Spark, filter and aggregate its datasets and provides interface for Apache Spark, DALEX (Descriptive mAchine Learning Explanations) - Illustrative set of tools that explicates working of complex models, Drake — Central R package that builds pipeline and checks for probability of reproducibility with dynamic computing power etc. These packages have substantial role in modern biological data analysis and interpretation of results in a very meaningful and comprehensible way for serving the masses. First of all, we are highlighting very basic problem in molecular biology that, how to apply χ2 test on sequence data using a human Zyxin gene with 2166 bp in length, and a null hypothesis of equal frequency of all nucleotides was postulated and tested. Similarly, Golub et. al gene expression data was also taken from the library “multtest” from bioconductor and analyzed on the basis of certain cut-off value. Then a frequency table was constructed showing true positive (tp), false positive (fp), true negative (tn) and false negative (fn), and a null hypothesis was formulated in both problems. χ2 test statistics was applied to check our hypothesis [1]. Furthermore, RNA sequencing and gene expression profiling workflow was also presented along with their allied set of packages being used for this sort of analysis. Different R packages are being used in NGS data analysis e.g. EdgeR, GEMMA, ggplot2, pdlyr, tidy, knitr, base and there are more than 10,000 packages available for almost all statistical methods.
Methods![]()
Installation of R and Bioconductor
R statistical libraries are installed from the link given on the site of bioconductor. Most libraries are pre-installed in R Platform but libraries/packages designed for specific tasks such as; statistical analysis, expression analysis, Z test etc., are need to installed from a designated online software portal named as “Bioconductor” https://www.bioconductor.org/install/ [2]. Bioconductor provides advanced statistical analysis techniques for life sciences owing to variety of libraries and progressively working algorithms.
Bioconductor covers widespread support for analysis of expression arrays, and well-developed support for exon, copy number, SNP, methylation and other assays. By using R platform for statistical analysis, bioconductor allied libraries concludes some important tasks such as pre-processing, quality assessment, differential expression, clustering and classification, gene set enrichment analysis and genetical genomics.
Bioconductor allied libraries also offers extensive interfaces to community resources to directly access large data sets from some renowned and important databases like NCBI-GEO, ArrayExpress, Biomart, genome browsers, GO, KEGG and diverse annotation sources.
R Methods
- Constructing Data Matrix
A matrix comprises 4 genes and fold change values of gene expression data from 3 individuals named Meerab, Safi and Kaif is constructed here by the following command [3, 4].
Gene1 <- c(1.00,1.50,1.25)
Gene2 <- c(1.35,1.55,1.00)
Gene3 < -c(-1.10,-1.50,-1.25)
Gene4 < -c(-1.20,-1.30,-1.00)
rowcolnames<- list(c("Gene1","Gene2”,”Gene3”,”Gene4”),+ c("Meerab","Safi","Kaif"))
gendat <-matrix(c(Gene1,Gene2,Gene3,Gene4), nrow=4, ncol=3, byrow=true, dimnames= rowcolnames) [5]
Data Matrix obtained from the above command by printing “gendat”
Meerab Safi Kaif
Gene1 1.00 1.50 1.25
Gene2 1.35 1.55 1.00
Gene3 -1.10 -1.50 -1.25
Gene4 -1.20 -1.30 -1.00
b. Computing on data matrix
To find out the mean following command was printed.
gendat is a variable that can be created by the user and we created above Data Matrix
Command: apply(gendat,1,mean) [6]
Gene1 Gene2 Gene3 Gene4
1.25000 1.300000 -1.283333 -1.166667
Command: apply(gendat,2,mean)
Meerab Safi Kaif
0.0125 0.0625 0.0000
c. Find Matrix row mean (largest to smallest)
meanexprsval <- apply(gendat,1,mean)
,- order(meanexpsval,decreasing=TRUE)
By printing “o” we will obtain the results.
[1] 2 1 4 3
d. Reordering the matrix
Command: gendat[o,]
Meerab Safi Kaif
Gene2 1.35 1.55 1.00
Gene1 1.00 1.50 1.25
Gene4 -1.20 -1.30 -1.00
Gene3 -1.10 -1.50 -1.25
To find out whether the row mean is positive or not
Command:
meanexprsval > 0 [7]
Following results were obtained.
Gene1 Gene2 Gene3 Gene4
TRUE TRUE FALSE FALSE
e. Computing of Golub Data Martix
Library(multtest); data(golub) (Golub et al., 1999) [8]
golub.gnames
A big data matrix was printed on the screen, showing the rows and columns.
Command: nrow(golub)
[1] 3051
Command: ncol(golub)
golub[,1]
[1] 38
Command: golub.gnames[1042,]
[1] "2354" "CCND3 Cyclin D3" "M92287_at"
Command: gol.fac <- factor(golub.cl, levels=0:1, labels = c("ALL","AML"))
Golub[1042,gol.fac==”ALL”] [9]
[1] 2.10892 1.52405 1.96403 2.33597 1.85111 1.99391 2.0597 1.81649 2.17622
[2] 1.80861 2.44562 1.90496 2.76610 1.32551 2.59385 1.92776 1.10546 1.27645
[3] 1.83051 1.78352 0.45827 2.18119 2.31428 1.99927 1.36844 2.37351 1.83485
meanALL <- apply(golub[,gol.fac==”ALL”], 1, mean)
meanALL
A large dataset was obtained in the result and not shown here to save space.
To find out the gene row
Print command:
Grep ("CD33",golub.gnames[,2])
Result obtained:
[1] 808
which explain that CD33 gene is located in 808 row of the golub data.
Chi-Square statistical testing on the Sequence data
Chi-square statistics are applied on the human Zyxin gene sequences extracted from NCBI (National Center for Biotechnology information), to test the hypothesis whether nucleotide frequency is equally distributed in these genes or not [10]. This was our null hypothesis, following commands were applied in R using the buit-in function of Chi-Square test and attaching the Zyxin gene sequences from NCBI. Pre-set and by default 0.05 significance level was used in this example.
https://www.ncbi.nlm.nih.gov/nuccore/X94991.1 [10].
Print Command:
Library (ape)
zyxinfreq <- table(read.genbank(c("X94991.1"),as.character=TRUE))
chisq.test(zyxinfreq)
Following results were obtained
Data: zyxinfreq
χ²-square = 187.0674, df = 3, p-value <2.2e-16
As the p value is less than significant level, so our null hypothesis was rejected and proved that the four nucleotides are not equally distributed in the given sequence.
Chi-Square statistical testing on gene expression data
After constructing the frequency table, following commands were printed to test our null hypothesis that whether a diseased state of a person is independent of gene expression cut-off values.
Print Command:
dat <- matrix(c(15,4,4,15),2,by row=TRUE)
chisq.test(dat)
Following results were obtained
Data: dat
χ²-square = 10.5263, df = 1, p-value =0.001177
So, the alternative hypothesis was accepted as p value was smaller than the significance level.
Next generation sequencing (NGS) data analysis
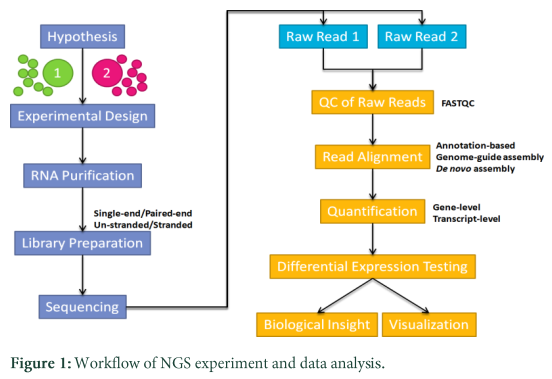
NGS data analysis is a technical and sensitive job as the analysis you perform through NGS platform is quite expensive and results are totally depending on its efficiency. R and its allied packages provide some efficient facilities for hypothesis testing whether it is whole genome sequencing, exome sequencing or whole transcriptome sequencing. Let’s take an overview of RNA Seq experiment and shed light that how many different steps are involved and how one can infer results from this experiment using R and its allied package libraries.
Differential expression analysis using whole transcriptome sequencing
Experimental Design
Proper experimental design is important to save resources. Experimental variations may be reduced by increasing the biological replicates, while comparison among different samples may also be accomplished by increasing the technical replicates [12]. Similarly, large number of samples helps a scientist to conclude statistically robust results while smaller sample size would not, but as an argument, sometimes reducing the number of individual allow to address the intra individual variabilities and control samples always help us to have an insight of interactions effects among case-control studies or to compare two different physiological conditions [13].
RNA Sequencing workflow
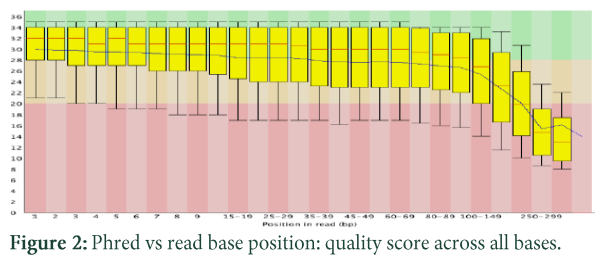
Normally pair end reads are obtained from the sequencer, in which total RNA is obtained from the source and transcribed into cDNA through reverse transcription. Then cDNA is fragmentized and adaptor sequences are ligated with these fragments, later on these fragments are fix to the surface and amplified through addition of bases. There are lot of technologies emerging all the time and it might be possible that one read will be as long as of a whole transcript [14]. Normally, fastq files are obtained, which have the read identifiers and Phred quality scores are assigned to strings of DNA, which gives probability of error, quality score and accuracy in percentages [13].
Introduction of R allied packages for RNA sequence Data analysis
As mentioned previously that Next Generation Sequencing is a very sensitive task to analyze so it needs proper computational algorithms which give a proper defined data flow for single task which is going to be analyzed further. In order to cope with such chaotic problems R Programming has different packages or libraries which multitasks according to the need of analysis. Here some of R related libraries are discussed and also their real time function are interpret in line by using RNA sequences to analyze NGS (next generation sequence) data.
- Library (affycoretools)
Libraries are an essential and integral part of R programming which allows to perform task with respect to the designed algorithms on which these libraries are based upon. Here some of most important and core libraries are used to analyze different NGS data sets which are used in many system biology and bioinformatics analysis tasks [15].
This library can be installed by the same R resource using the command in R prompt:
("https://bioconductor.org/biocLite.R")
biocLite (“affycoretools”) [16].
- Library(edgeR)
edgeR is the most important library used for differential gene expression analysis specially on RNA-seq data. edgeR is based on negative binomial distribution which further evaluates statistical analysis for disease diagnosis. Some other major tasks done by this library are empirical Bayes estimation, exact tests, generalized linear models and quasi-likelihood tests. edgeR is a multitasking library which not only use for these above-mentioned tasks, but also in many core computational and system biology data analysis. Such as, for finding gene pathways, data normalization, differential methylation, geneSet enrichment scores.
This package is installed by using a command;
biocLite(“edgeR”)
initialization command library(edgeR)
- Library(limma)
Library limma is an associated package with edgeR. Moreover, it is used to perform some additional tasks like, statistical, linear and differential expression analysis on microarray data. Library (limma) also runs with the same set of commands [17].
biocLite(“limma”)
Besides these, some other libraries are also used for same purpose such as DEGseq and NOIseq.
RNA-seq data analysis
Recent technological developments acquaint with high-throughput sequencing approaches. A variability of experimental procedures and analysis are summarized for gene expression, regulation and encoding of genetic variants [18].
In this article differential gene expression analysis is done on a large dataset of non-castrated prostate cancer treated with docetaxel chemotherapy and expression profiling is further done on all 6 pre and post treatment patient. Raw data of cancer patient was extracted from NCBI-GEO (gene expression omnibus) database against gene ID: GSE51005. After the extraction of raw data file and converted into CSV (comma separated values file) further analysis is done with the help of R programming allied datasets for differential gene expression analysis, are used to read Raw data file of GEO database and then data is explored by using libraries (affycoretools) different plots are visualized using raw data file based on RNA read counts such as; Boxplots and Densityplots [20]. After that libraries (Limma and edgeR) are initialized for data normalization and obtain RPKM (Reads Per Kilo-base Per Million Mapped Reads) values. RPKM is a method for computing the gene expression from RNA sequencing data by normalizing total read length and also the number of sequencing reads [21, 22].
Normalization of the data obtained from the GEO database of pre-and post- treatments of Prostate cancer patients by using the R packages (using Limma , RPKM values) to analyze differentially expressed genes which were mainly involved in causing Prostate cancer and found the up- and down- regulatory genes associated with these causing [23].
R commands for above mentioned analysis;
Raw Data reads:
raw.data<- read.csv("GSE51005_rawData_file.csv",header=T, row.names=1)
Data normalization is used to find up and down regulation of genes involved in non-castrated prostate cancer, which is further utilized to make gene expression pathways of respected genes. After finding up and down regulatory genes using R (limma library) data is downloaded in CSV format and further analysis is done by using different gene annotation and pathway analysis tools.
Results![]()
Data exploration
- Boxplots
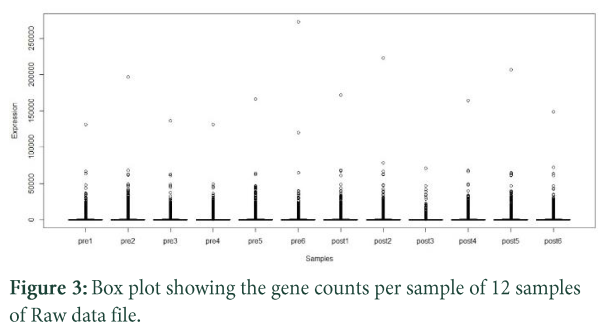
RNASeq counts data is very diverse from continuous microarray data so it is better to do some basic exploration of the data to find variation in the data using boxplots generation code. Due to large dataset it is quite unprecise to evaluate variation between pre and post treatment of patients [24]. Then we use log2 scale for transforming data into much aligned and precise manner for further normalization. A detailed boxplot results of normalized data is shown in (Figure 3).
R code for boxplots using log2 scale:
- boxplot(log2(raw.data[,1:12]+0.01), col=rep(c("green","red"), each=3), xlab= "Samples", ylab="Raw Expression in log2 scale")
- Density Plots
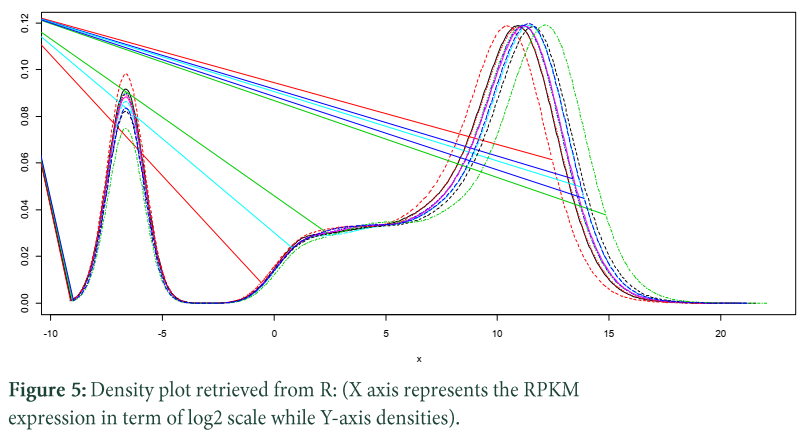
Density plot illustrates the genes that are extremely expressed on highest RPKM. The expression of plot ranges from 20 (1 RPKM) to 220 (1048576) RPKM, which seems like a bimodal distribution but the first mode before zero is considered as neglected because we have added a small constant to null values [25].
R command for computing density plots:
plotDensity(log2(raw.data[,1:13]+0.01))
formula used to find RPKM values:
raw.data[,2+i] / (raw.data$Length/1000) / (library.sizes[i]/1000000)
Clustering and separation
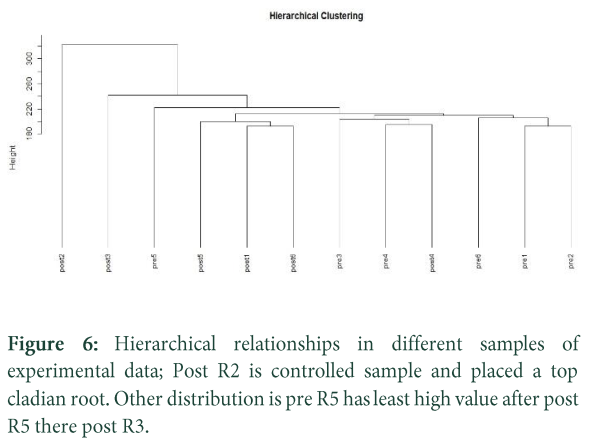
Initial cluster analysis is done to see how similar the samples are after normalizing data. Fast hierarchical clustering is done by with the help of WGCNA package (Weighted Correlation Network Analysis), which is useful for predicting outliers in any major groupings of samples. Clustering on the RPKM values are done, but again they need to have a small constant added and the log2 taken [25]. Then to perform clustering of samples, data matrix must be transposed so that the rows X columns are samples X Genes. Finally, we calculate a distance matrix between the samples and perform the hierarchical clustering for further analysis.
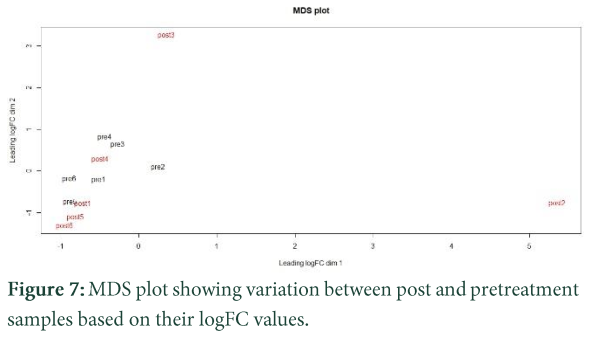
MDS plots
MDS (multidimensional scaling) delivers a visual representation of propinquities amongst the set of entities. In MDS plots those sequences which are similar to each other are showed at same location and those which have variants will be apart. From the above figure, we observe that sample 3 and sample 2 (tax.Post.R3 and tax.Post.R2) is showing more variance. Then further normalization is done to remove the zero counts from our data which may influence our results [21]. After this, further analysis is predicted by using edgeR package for finding up and down regulatory genes involving in disease pathways. These genes are further analyzed and pathways are predicted with the help of other gene annotations and functional tools.
Figures & Tables
Discussion![]()
A large dataset of prostate cancer pateints was taken from a well known database NCBI-GEO (Gene expression Omnibus). This data was based on experimentation of chemotherapic drug resistance between 6 individuals in which half of them are newly dignosed prostate cancer while others have non-castration resistant prostate cancer. On the basis of this successful experiment, we decided to analyze some in silico differential gene expression analysis. For that purpose R programming is used to produce different statistical analysis on given data. Assorted R packages are used which are basically designed for differential expression analysis, some major packages among them are limma, edgeR, DEseq [2].
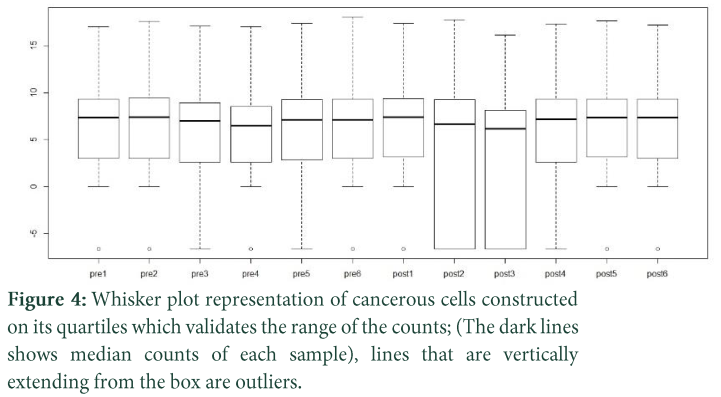
Various dataplots are constructed which makes data more precise and accurate. RPKM (Reads Per Kilo-base Per Million Mapped Reads) values are calculated, barplots and whisker plots are generated on the basis of these values. Zero read counts are eliminated for normalization. Hierarchical clustering is constructed to see the variation between post and pre treatment of pateints, up and down regulation of genes are extracted from data to make gene pathways.
Major concept for this insilico research is based around predictive and personalized medicine, while studying gene pathways and their mechanism in our body different dose of medicine is given according to response towards that drug from our body. If a pateints body is responding in a very effiecient manner then dosage of drugs will be regulated according to improvement, and if body showed negative efficiency towards medicine then further analysis is preformed to check major causes and after that maximum progression for cure will be designed. Predictive medicine is also another major field in which initial genetic screening is done to check reason of development of disease. When respective genes were extracted from data it is further analysed for the purpose to find disease related genes, their mechanism and their resistance if any with drugs. Another major benefit of personalized medicine is that one can design a specific drug which is only used on individual for whom it will be customized.
The authors declare that there is no conflict of interest regarding the publication of this paper.
References![]()
- Anderson DR, Burnham KP, Thompson WL. Null hypothesis testing: problems, prevalence, and an alternative. The journal of wildlife management, (2000); 912-923.
- Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S. Bioinformatics and computational biology solutions using R and Bioconductor. Chapter: Book Name. 2006 of publication; Springer Science & Business Media.
- Gentleman R. R Programming for Bioinformatics. 2008; 53: 4200‐6367. Chapman & Hall/CRC
- Hartigan JA. Direct clustering of a data matrix. Journal of the american statistical association, (1972); 67(337): 123-129.
- Krijnen WP. Applied statistics for bioinformatics using R. Institute for Life Science and Technology, Hanze University, (2009).
- Mirsky E, DeHon A. MATRIX: a reconfigurable computing architecture with configurable instruction distribution and deployable resources. In FCCM, (1996); 96: 17-19.
- Pinar A, Heath MT. Improving performance of sparse matrix-vector multiplication; 1999. ACM. pp. 30.
- Quandt K, Frech K, Karas H, Wingender E, Werner T. Matlnd and Matlnspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic acids research, (1995); 23(23): 4878-4884.
- Elmroth E, Gustavson F, Jonsson I, Kågström B. Recursive blocked algorithms and hybrid data structures for dense matrix library software. SIAM review, (2004); 46(1): 3-45.
- Tallarida RJ, Murray RB (1987) Chi-square test. Manual of Pharmacologic Calculations: Springer. pp. 140-142.
- Wilcox RR Introduction to robust estimation and hypothesis testing. Chapter: Book Name. 2011 of publication; Academic press.
- Getts RC, Kadushin J (2016) Whole transcriptome sequencing. Google Patents.
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nature methods, (2009); 6(5): 377.
- Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nature Reviews Genetics, (2016); 17(6): 333-351.
- MacDonald JW, MacDonald MJW, biocViews ReportWriting M, OneChannel G. Package ‘affycoretools’.
- Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics, (2013); 29(14): 1830-1831.
- Wettenhall JM, Smyth GK. limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics, (2004); 20(18): 3705-3706.
- Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics, (2009); 26(1): 136-138.
- Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, et al. A survey of best practices for RNA-seq data analysis. Genome biology, (2016); 17(1): 13.
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews genetics, (2009); 10(1): 57.
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, (2010); 26(1): 139-140.
- Rapaport F, Khanin R, Liang Y, Pirun M, Krek A, et al. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome biology, (2013); 14(9): 3158.
- Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome biology, (2010); 11(3): R25.
- Spitzer M, Wildenhain J, Rappsilber J, Tyers M. BoxPlotR: a web tool for generation of box plots. Nature methods, (2014); 11(2): 121.
- Wilson PW. FEAR: A software package for frontier efficiency analysis with R. Socio-economic planning sciences, (2008); 42(4): 247-254.
![]()
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0