

Full Length Research Article
Genetic Analysis of SCN5A Gene in Patients of Long QT Syndrome
Uzma Zaheen1, Memona Yasmin1, Saadia Noreen1, Saqib Ali2*, Allah Rakha1
Adv. life sci., vol. 7, no. 1, pp. 27-31, November 2019
*– Corresponding Author: Saqib Ali (Email: jawadfs@hotmail.com)
Authors' Affiliations
2. University of Agriculture, Faisalabad, Pakistan
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Finding out the cause of death is the main concern in forensic casework, particularly in sudden unexplained death cases. Long QT syndrome is a form of life debilitating cardiac arrhythmia. Among LQT genes, LQT3 is suggested to be more lethal and patients are at high probability of sudden death as compared to LQT1 and LQT2 patient.
Methods: Samples of long QT patients were collected and the coding regions of all exons of the SCN5A, voltage- gated Na+ channel gene were screened by sequencing for the potential mutations as the causative agent of long QT syndrome.
Results: After data analysis, 3 genetic variations have been found, amongst them are 2 heterozygous mutations that were reported previously in other ethnicity G87A-A29A found in exon 2, G1673A-H558R in exon 12 and one novel mutation that has not been reported so far, G>A 1238-G412G resulting in a transition mutation and change in amino acid GGG- Glycine to GAG- glutamic acid at position 412 in exon 9. While in other exons, no significant mutation was found.
Conclusion: As some of the exons showed mutation and the sample size was small, however, further functional analysis of the gene is needed with large number of samples for the confirmation of results.
Keywords: Long QT Syndrome; SCN5A gene; Genetic variations; Sudden death
Introduction![]()
In the developed states, sudden cardiac death (SCD) is the commonest cause of death, where about 70-80 % deaths are due to cardiac abnormalities [1]. Long QT syndrome is one of the main causes of SCD. It is very rare with a prevalence of 1 in 10,000 to 15,000 individuals. Pathogenic variants causing Long QT syndrome has been identified in 13 genes such as LQT1 (KCNQ1), LQT2 (KCNH1), LQT3 (SCN5A), KCNE1, KCNJ2, KCNJ5, RYR2, SCN4B, ANK2, CACNAK, CAV3 and KCNE2 [2]. The pathogenic mutations in LQT1, LQT2 & LQT3 are responsible for 80% of long QT syndrome [3].
A significant number of deaths, particularly in the young, are due to inherited heart disorders, both for structural and functional heart disease [4]. In recent decades, a large number of developments have been done to understand the genetic cause of SCD [5]. It is important to rule out the genetic cause of sudden death being the reason that other family members are also in danger of having a fatal cardiac condition. Recent studies show a rate of 30–200/100,000 individuals every year [6], bringing about around 335,000 passing every year in the USA. Similarly, in Europe, the rate is obviously higher in Northern nations. There are many possible factors that could lead to the sudden cardiac death like hypertrophic cardiomyopathy, coronary artery abnormalities and long QT syndrome. Post-mortem genetic estimation, or “molecular autopsy” is becoming progressively significant in the evaluation of sudden death cases, mainly for identification of pathogenic alterations in genes underlying disease [7]. This strategy may have an important effect on the screening of family members at risk, but still it is not a routine component of post-mortem testing [8].
Association of several genes with LQT is reported previously like LQT1 (KCNQ1), LQT2 (KCNH2), LQT3 (SCN5A), KCNE1, KCNJ2, KCNJ5 and KCNE2. The LQT1 gene, also known as KCNQ1, accounts for 30-35% of all LQT cases. It is positioned at chromosome 11p15.5 and encodes voltage-gated potassium channels. LQT2 form is the second common gene, situated on chromosome 7, accounts for 25 to 30% of all LQT cases [9]. The third gene responsible for the LQT syndrome is LQT3 gene, also known as SCN5A, accounts for 5-10% of all LQT cases [10]. The sodium channel gene is situated on 3rd chromosome p21, it consists of 28 exons. It encodes protein of 2,016 amino acids [11].
SCN5A, voltage-gated sodium channel gene. It is dominantly expressed in the heart [12]. The mutations in SCN5A slow down or inactivate the sodium channel, as a result delaying in depolarization. The persistent late Na+ current extends the plateau phase of action potential and causes long QT segment in ECG. Variants related to SCN5A gene encodes the α-subunit of the cardiac sodium channel (Nav1.5), results in a range of disease individuals, labeled as sodium channelopathies [11].
Mutations in SCN5A gene are known to cause sudden unexpected death syndrome, a situation frequently encounter in forensic science [13]. In present study, the mutant variant of SCN5A gene in LQT syndrome patients has been identified. Subsequently, this study could be extended to ‘molecular autopsy’ for the doctors to diagnose the LQT syndrome by the help of genetic testing in sudden death cases to resolve the query of unexplained and unexpected sudden deaths.
Methods![]()
Sample Collection
Six samples were collected after complete evaluation of patients from two Hospitals of Lahore, Pakistan. The patients of congenital Long QT Syndrome diagnosed by cardiologist were ascertained from pediatric cardiology ward, Children Hospital Lahore, Pakistan and cardiology department, PIC Lahore, Pakistan. The patients of Long QT were selected on the basis of Schwartz’s LQTS, Diagnostic criteria (2006) [14]. An informed consent was obtained from the affected individuals in written form. The consent for patient below 12 year was taken from his/her parents. The blood samples were taken in EDTA containing vacutainers and stored at 4oC for further use. The relevant clinical data i.e. age, family history, ECG records and other related health information was recorded. The confidentiality of the data has been maintained in strict accordance with the protocol approved by ethical committee of the institute.
Extraction and Quantification of DNA
The Phenol Chloroform Isoamyl alcohol method was used for DNA extraction [15]. The quality and quantity of the extracted DNA was estimated by using agarose gel electrophoresis (0.8%) and gel documentation system (Bio Red System) (Fig. SF1, SF2).
Primer Designing
The primers used for this work were retrieved from the literature as reported by Park et al., [11] (Table ST1).
Primer Optimization
The primers for all the exons were optimized via gradient PCR (Figure SF3).
DNA Amplification
The DNA regions of selected exons of SCN5A were amplified by using conventional PCR (Table ST2, Figure SF1).
Resolution of PCR product on gel
The size of the PCR products of all the exons were visually examined by resolving the amplicons on agarose gel (Figure SF4, SF5).
The amplified DNA was sequenced, and the data was analyzed by using Chromas Lite v2.01 software. The sequence of each exon was blast against the normal reference sequence of SCN5A by using BLAST (Basic Local Alignment Search Tool) sequence server on NCBI website.
Results![]()
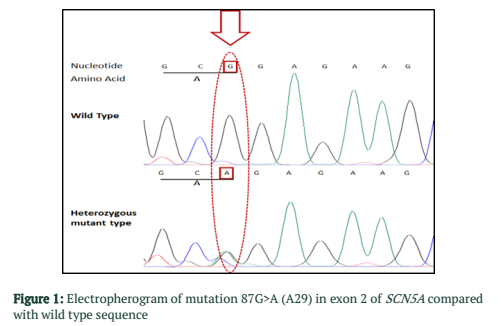
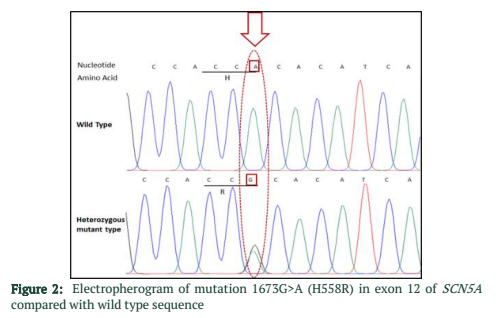
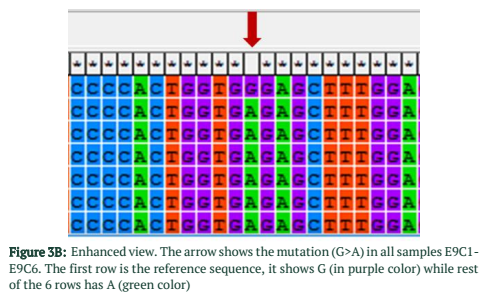
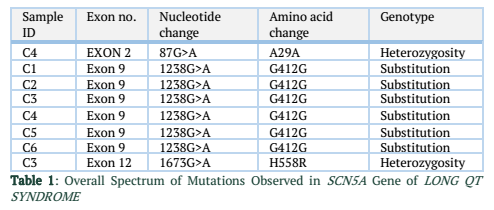
The exons of SCN5A gene were sequenced of six samples. After the sequence analysis, three genetic variations were identified, two of them are heterozygous type and one is missense mutation. One of the heterozygous mutations (G87A-A29A) was found in exon 2 of sample C4 (E2 C4) (Figure 1), there is heterozygosity of G and A at position 87 resulting GCG to GCA both are alanine at position 29. The other heterozygous mutation (G1673A-H558R) is 1673G>A in exon 12 of sample C3 (E12 C3) (Figure 2) resulting in a change of amino acid to CAC-Histidine (H) to CGC- Arginine(R) at position 558. The third mutation (missense) was identified in exon 9, where a substitution of G to A at 1238 position (G>A 1238-G412G) was detected and change in amino acid GGG- Glycine to GAG- glutamic acid at position 412 (Figure 3A, 3B). The transition mutation was found in all six samples (C1-C6) (Figure SF4, SF5) (Table 1). For the verification of mutational analysis, the sequenced data was compared with the Punjabi population data that is available on 1000 Genomes. The comparative analysis showed that no mutation (87G>A-A29A, 1238G>A-G412G, 1673G>A-H558R) was detected in the control data (Table 1).
Figures & Tables

Discussion![]()
Several SCN5A variations have been reported in other countries. These variations are associated with different cardiac diseases like LQTS, Brs, AV block, conduction defect, AF, VF, and overlapping syndrome [16]. Few studies has been reported so far, to find out the genetic variations of SCN5A gene with reference to Long QT Syndrome, although previously many studies related to SCN5A variation associated with various cardiac disease like Bruganda syndrome, progressive cardiac conduction defect, AF, AV conduction block and overlapping syndrome [17]. Park et al., in 2012 worked on Korean population and found mutational change in exon 2, 12, 17, 20, 28 of SCN5A gene [11]. Splawski et al., in 2000 found that most of the mutations in SCN5A gene were located in exon 23 to 28 [18]. Iwasa et al., in 2000 checked the polymorphism of LQTS genes in Japanese population; they reported 20 SNPS in 4 genes of LQTS and their allelic frequencies, 7 polymorphism detected in LQT1, 6 in LQT2, 5 in SCN5A and 2 in KCNEI [19]. They found polymorphism in SCN5A at exon 28, 12, 18, 24, and 28. Keller et al., in 2003 found the novel mutation in SCN5A gene located at exon 26 [20]. Wang et al., in 2005 observed that a SNP R1193Q of the SCN5A gene is noted in few overall communities, although its occurrence fluctuates in different ethnic foundation with frequency of 0.2% to 12%. Characterization of ECG findings revealed that individuals having R1193Q are at expanded danger of causing LQTS [21].
According to a study conducted in Korean population 2012, a number of SCN5A variations were found in AV block patients [11]. The mutations, G87A-A29A and IVS9-3C > A, identified through this study had already been reported in West literature, while T5457C- D1819D SNP has been reported in Asian literature [22]. A1673G-H558R is situated on sodium channel, and past studies revealed that this H558R variant can alter the phenotype of disease-producing SCN5A mutations. A1673G-H558R was notorious in 18% of USA population, 8% of Japanese communities. G87A-A29A a common SNP found in Han Chinese population [11].
H558R variant causes change in amino acid in sodium channel gene. It is a common polymorphism with frequency of 20–30% in Caucasian people. The single change in amino acid didn’t produce any symptoms, but it can help to produce lethal changes in SCN5A gene [23].
In 2014, Boehringer et al., conducted a study, according to him 4 variants (87G>A, 1673A>G, IVS16-6C>T and 5457T>A) determined common variants of which only 1673A>G (His558Arg) was a missense variant because in which there is change in amino acid [24]. It was found that the two SCN5A variants G87A-A29A and G1673A-H558R are also present in local Pakistani population. Along with that a novel variant G>A 1238-G412G was also identified.
In conclusion, the 3 important genetic variants of SCN5A gene have been found. However, a functional analysis of the mutation is also suggested to confirm the pathogenicity of the disease.
Authors' Contribution
Uzma Zaheen: Designed the study, carried out the genomic work. Memona Yasmin: Participated in sample collection. Saadia Noreen: Performed data analysis. Saqib Ali: Grammar checking of the manuscript. Allah Rakha: Principle Investigator of the research.
The authors declare that there is no conflict of interest regarding the publication of this paper.
References![]()
- Rodríguez-Calvo MS, Brion M, Allegue C, Concheiro L, Carracedo A. Molecular genetics of sudden cardiac death. Forensic science international, (2008); 182(1-3): 1-12.
- Stattin E-L, Westin IM, Cederquist K, Jonasson J, Jonsson B-A, et al. Genetic screening in sudden cardiac death in the young can save future lives. International journal of legal medicine, (2016); 130(1): 59-66.
- Zipes DP, Wellens HJ. Sudden cardiac death. Circulation, (1998); 98(21): 2334-2351.
- Schwartz PJ, Ackerman MJ. The long QT syndrome: a transatlantic clinical approach to diagnosis and therapy. European heart journal, (2013); 34(40): 3109-3116.
- Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation, (2001); 103(1): 89-95.
- Virmani R, Burke AP, Farb A. Sudden cardiac death. Cardiovascular pathology, (2001); 10(5): 211-218.
- Liu C, Zhao Q, Su T, Tang S, Lv G, et al. Postmortem molecular analysis of KCNQ1, KCNH2, KCNE1 and KCNE2 genes in sudden unexplained nocturnal death syndrome in the Chinese Han population. Forensic science international, (2013); 231(1-3): 82-87.
- Kauferstein S, Kiehne N, Jenewein T, Biel S, Kopp M, et al. Genetic analysis of sudden unexplained death: a multidisciplinary approach. Forensic science international, (2013); 229(1): 122-127.
- Shinnawi R, Gepstein L. iPCS cell modeling of inherited cardiac arrhythmias. Current treatment options in cardiovascular medicine, (2014); 16(9): 1-18.
- Ackerman MJ, Splawski I, Makielski JC, Tester DJ, Will ML, et al. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm, (2004); 1(5): 600-607.
- Park HS, Kim YN, Lee YS, Jung BC, Lee SH, et al. Genetic analysis of SCN5A in Korean patients associated with atrioventricular conduction block. Genomics & informatics, (2012); 10(2): 110-116.
- Grant AO. Molecular biology of sodium channels and their role in cardiac arrhythmias. The American journal of medicine, (2001); 110(4): 296-305.
- Vatta M, Dumaine R, Varghese G, Richard TA, Shimizu W, et al. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Human molecular genetics, (2002); 11(3): 337-345.
- Schwartz PJ. The congenital long QT syndromes from genotype to phenotype: clinical implications. Journal of internal medicine, (2006); 259(1): 39-47.
- Green M, Sambrook J, Sambrook J. Molecular cloning: a laboratory manual 4 edition Cold Spring Harbor Laboratory Press. New York, (2012).
- Koo S, Teo W, Ching C, Chan S, Lee EJ. Mutation screening in KCNQ1, HERG, KCNE1, KCNE2 and SCN5A genes in a long QT syndrome family. ANNALS-ACADEMY OF MEDICINE SINGAPORE, (2007); 36(6): 394.
- Veltmann C, Barajas‐Martinez H, Wolpert C, Borggrefe M, Schimpf R, et al. Further insights in the most common SCN5A mutation causing overlapping phenotype of long QT syndrome, Brugada syndrome, and conduction defect. Journal of the American Heart Association, (2016); 5(7): e003379.
- Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, et al. Spectrum of mutations in long-QT syndrome genes. Circulation, (2000); 102(10): 1178-1185.
- Iwasa H, Itoh T, Nagai R, Nakamura Y, Tanaka T. Twenty single nucleotide polymorphisms (SNPs) and their allelic frequencies in four genes that are responsible for familial long QT syndrome in the Japanese population. Journal of human genetics, (2000); 45(3): 182-183.
- Keller DI, Acharfi S, Delacrétaz E, Benammar N, Rotter M, et al. A novel mutation in SCN5A, delQKP 1507–1509, causing long QT syndrome:: Role of Q1507 residue in sodium channel inactivation. Journal of molecular and cellular cardiology, (2003); 35(12): 1513-1521.
- Wang Q. Author’s reply: link of SCN5A SNP R1193Q to long QT syndrome. Journal of medical genetics, (2005); 42(2): e8-e8.
- Gellens ME, George AL, Chen L, Chahine M, Horn R, et al. Primary structure and functional expression of the human cardiactetrodotoxin-insensitive voltage-dependent sodium channel. Proceedings of the National Academy of Sciences, (1992); 89(2): 554-558.
- Viswanathan PC, Benson DW, Balser JR. A common SCN5A polymorphism modulates the biophysical effects of an SCN5A mutation. The Journal of clinical investigation, (2003); 111(3): 341-346.
- Boehringer T, Bugert P, Borggrefe M, Elmas E. SCN5A mutations and polymorphisms in patients with ventricular fibrillation during acute myocardial infarction. Molecular medicine reports, (2014); 10(4): 2039-2044.
![]()
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0