

Full Length Research Article
Evaluation of a recurrent mutation in HGF gene responsible for non-syndromic hereditary deafness in Kashmiri population
Kalsoom Zaigham1*, Hamna Tariq1, Tanveer Ahmed Qaiser2, Saad Bin Maqsood1, Asma Ali Khan1
Adv. life sci., vol. 7, no. 4, pp. 281-286, August 2020
*- Corresponding Author: Kalsoom Zaigham (Email: kalsimbb@yahoo.com)
Authors' Affiliations
2. Shaheed Zulfiqar Ali Bhutto Medical University Islamabad – Pakistan
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Mutations in gene coding for hepatocyte growth factor protein, HGF are responsible for hereditary deafness worldwide. Evaluation of recurrent variations displays prevalent heredity diversity of a specific population. Mutational screening of HGF was aimed to ascertain the causative recurrent variations in Kashmiri families.
Methods: Kashmiri families were enrolled from different divisions of Azad Jammu and Kashmir. By employing linkage analysis all the families were screened for loci common in Pakistani population. Families linked with DFNB39 locus were subjected to direct sequencing for mutational analysis of variants prevalent in Pakistani population.
Results: Sanger sequencing identified a noncoding c.482+1986_1988delTGA variant of HGF as recurrent mutation in Kashmiri population. These findings implicate this HGF variant as major contributing variant of hearing impairment in Kashmiri families with a frequency of 8.8%.
Conclusion: This is the first study conducted to elucidate the founder effect and prevalence of HGF variants in Kashmiri population. This study increases the prevalence of HGF variants associated with hearing impairment in the Kashmiri families.
Keywords: HGF; Autosomal recessive hearing loss; Recurrent mutations; Founder effect
Introduction![]()
Hearing impairment (HI) is a heterogeneous infirmity worldwide. In Pakistan incidence of bilateral HI is 1.6 per 1000 individuals [1-3]. The Kashmiri population of Pakistan is an invaluable resource to study the recessive mode of genetic disorders like HI due to high incidence rates of consanguineous marriages or mating within the same ethnic groups [4-6]. The hearing loss related gene mutation patterns vary significantly among different races and regions. In Kashmiri population almost 70% of the HI appears due to consanguineous reunions, consequently as compared to other populations of the world, the number of hearing impaired families in this population is much higher [7].
Deafness is caused by hundreds of genes that when get mutated cause pathogenicity [8-12]. Of the 93 non-syndromic hearing deafness (NSHD) loci that recorded so far, the pathogenic variations have been known for 75 genes (Hereditary Hearing Loss Homepage: https://hereditaryhearingloss.org/). This varies from cochlear specialized genes performing inner ear specific functions to housekeeping genes with ubiquitous expression [13,14]. For many monogenic disorders extensive genetic analysis studies have presented variations in noncoding part of the genes. One such gene, hepatocyte growth factor gene HGF has been reported to cause non syndromic deafness in large cohort of hearing impaired families due to variations present in noncoding region of the gene [15].
Human hereditary deafness (DFNB39) is concomitant with noncoding variations in the 3'UTR and intron of an isoform of HGF. The growth factors like hepatocyte growth factors (HGF) are essential for the development of auditory system and for the maintenance of hearing process in humans. In many different tissues this growth factor is involved to perform intricate signal transduction mechanism. Mouse model studies revealed abnormal cochlear tissue growth in inner ear in the absence of HGF. HGF is essential for integration of sensory crest cells into the central stria vascularis layer during the development of inner ear of mice. The stria retains an endocochlear potential (+80 to +120 mV) and high potassium concentration necessary for mechanotransduction of sound by inner hair cells of ear [16]. Using a HGF mutant mouse in a study [15] with a ten base pair deletion recapitulating a human DFNB39 noncoding variant. The findings of the study demonstrated failure of migration of neural crest cells to stria vascularis middle layer causing significant reduced endocochlear potential in inner ear hair cells.
Regulatory putative HGF variants have been reported to cause surprisingly a specific phenotype, non-syndromic deafness. HGF levels must be fine-tuned for normal hearing as an over or less HGF expression results in loss of hearing. In this study we identified one regulatory variant (c.482+1986_1988delTGA) in the non-coding region (intron 4) of the HGF as recurrent mutation in four Kashmiri families of Pakistan.
Methods![]()
An approval from Institutional Review Board was obtained for this study from National Centre of Excellence in Molecular Biology, University of the Punjab Lahore Pakistan. A total of 45 families with three or more affected individuals were visited and collected from different divisions of Azad Jammu and Kashmir region of Pakistan. Pedigrees were constructed for the confirmation of inheritance pattern and initial medical information. Detailed medical histories were recorded to rule out environmental factors and any associated syndromes.10 ml of blood sample was taken from all the participants of the study. Blood samples were preserved at -20 ºC for long term storage before DNA extraction.
Extraction and Quantification of Genomic DNA
For the extraction of DNA standard protocol of DNA extraction was performed [17]. The integrity and quantification of DNA was estimated by using Agarose gel electrophoresis.
Screening of Common Deafness Loci
Linkage analysis was performed initially for all the families to screen common prevalent deafness loci (DFNB1, DFNB2, DFNB3, DFNB4, DFNB12 and DFNB39) by using STR markers (fluorescently labeled). The pooled PCR products were evaluated on 3130 DNA genetic analyzer. Gene mapper software was used for allele calling. Cyrillic software was used for constructing pedigrees and Haplotypes. LOD (logarithm of odds) score was calculated by FASTLINK [18].
Mutational Screening
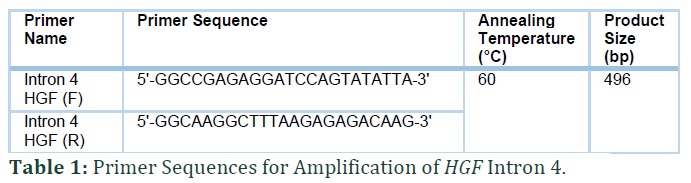
PCR (polymerase chain reaction) amplification following Sanger sequencing was done for one primer pair of HGF with most common non coding mutation in intron 4 (Table 1). The sequencing results were analyzed with Sequencher® 5.4.6 software (Gene Codes Corporation).
Results![]()
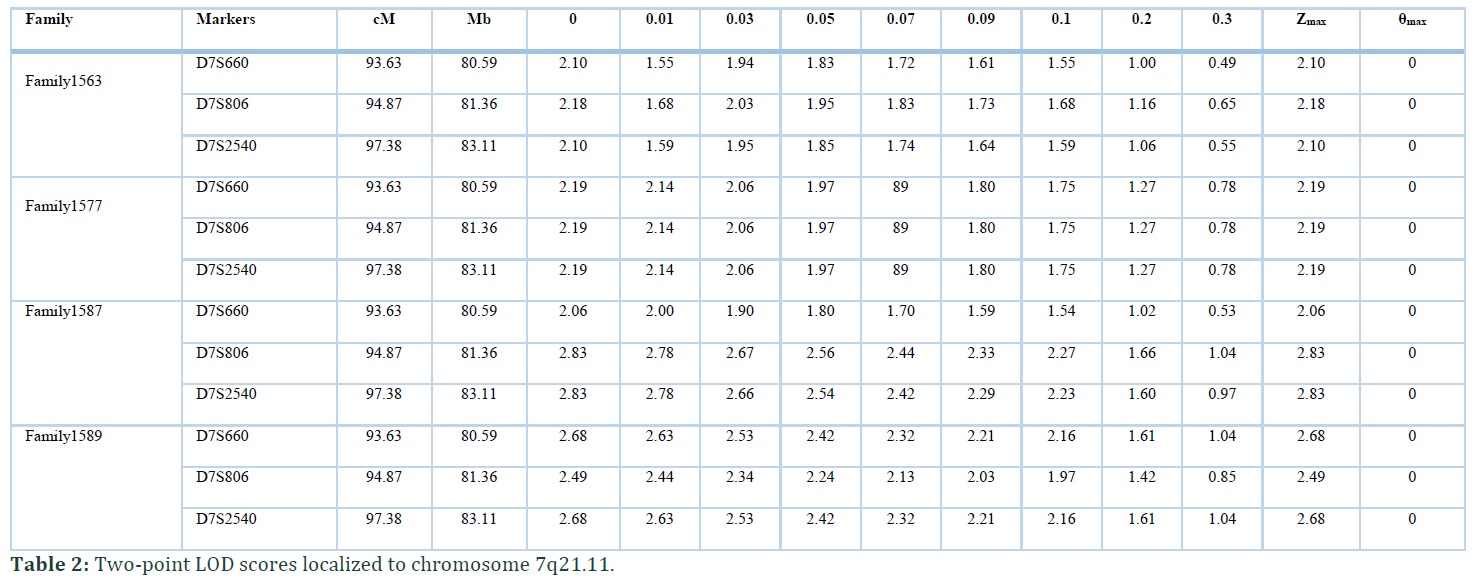
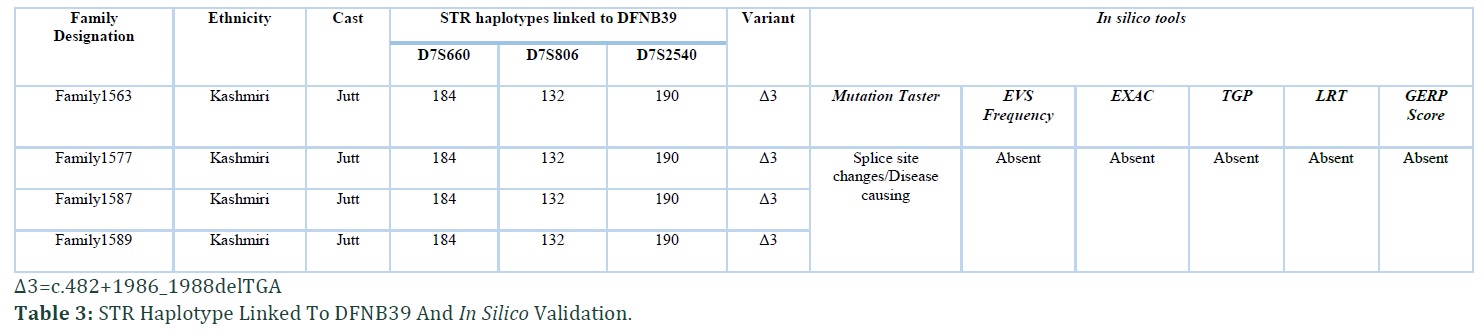
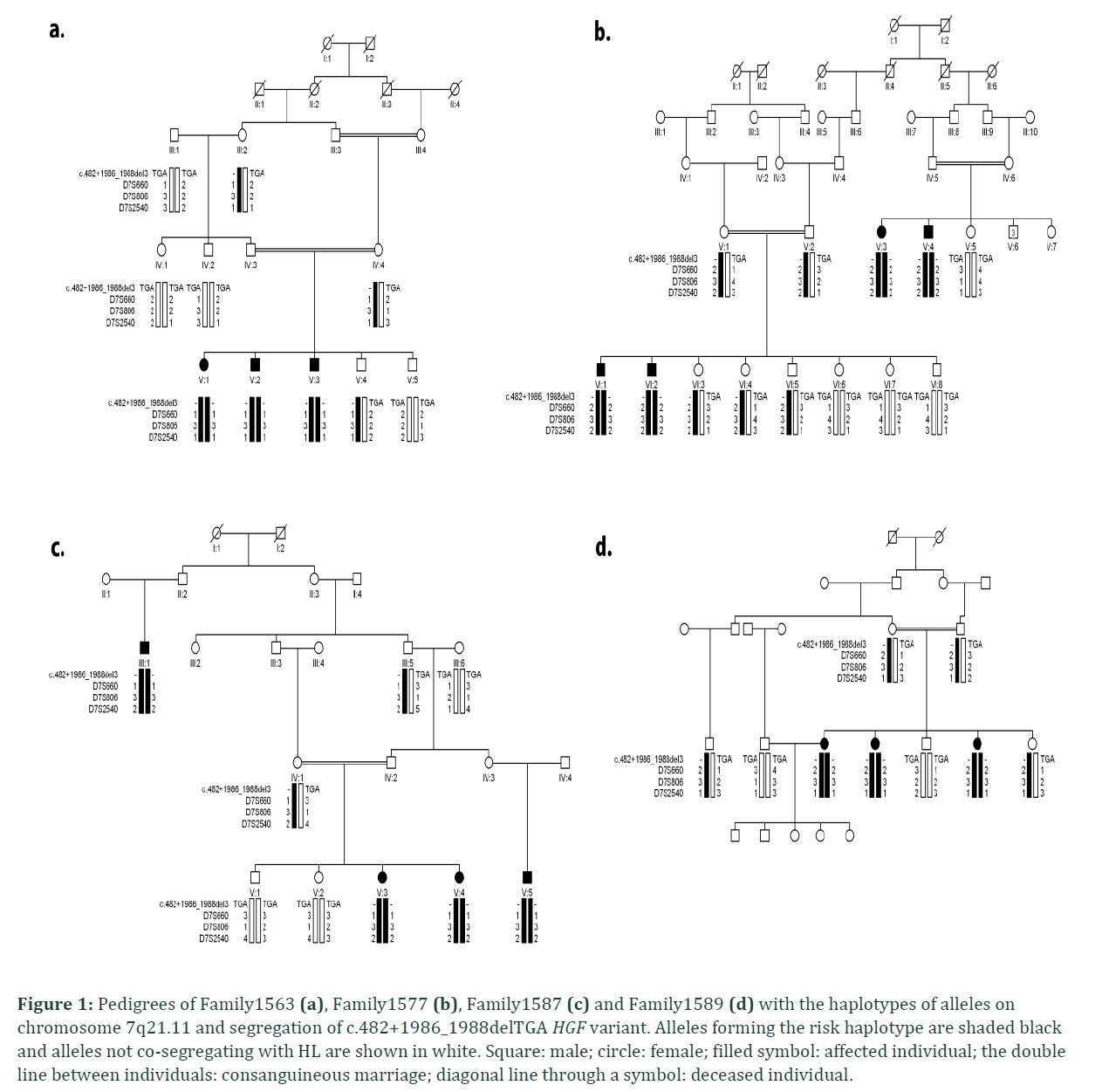
In the present study four consanguineous Kashmiri kindred, Family1563, Family1577, Family1587 and Family1589 with hearing impairment, were linked to DFNB39 locus with recurrent noncoding mutation in intron 4 of HGF. The families belonged to same ethnic (Kashmiri) background and caste (Jutt). Autosomal recessive inheritance pattern was found in all the four families (Figure 1). No affected individual from four families had complaint of night blindness or any other clinical manifestation. The haplotype of all hearing impaired individuals of families showed linkage to DFNB39 locus at chromosome 7q21.11. In Family1563 a maximum two point LOD score (Zmax) of 2.10 at θ= 0 was observed with marker D7S660 and D7S2540. Linkage was sustained to this region at distal (D7S660) and proximal side (D7S2540) for other three families with maximum two point LOD score (Zmax) of 2.19 (with markers D7S660, D7S806 and D7S2540), 2.83 (with markers D7S806 and D7S2540) and 2.68 (with markers D7S660 and D7S2540) at θ= 0 for Family 1577, Family 1587 and Family 1589 respectively (Table 2). Zmax was obtained with D7S2540 in all four families. The assembled haplotype also displayed same genotype pattern i.e., 184, 132 and 190 (Table 3) for all the four families revealing founder effect.
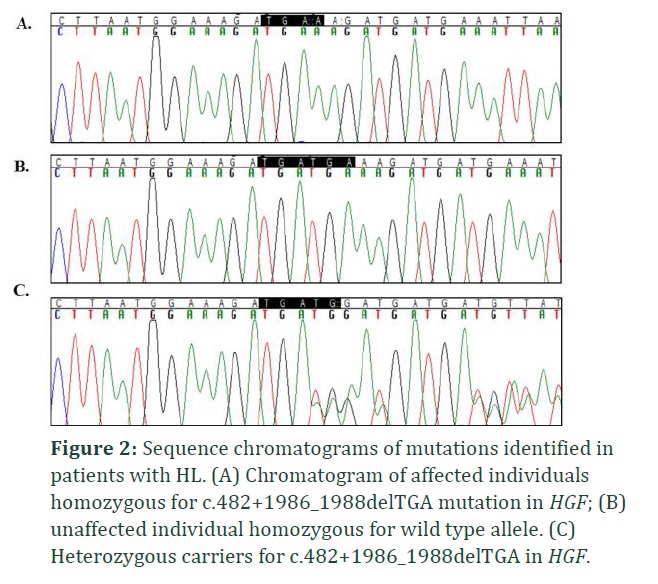
Bidirectional Sanger sequencing revealed that all the four families harbored common recurrent c.482+1986_1988delTGA mutation in intron 4 of HGF. All the affected individuals showed homozygosity for the identified mutation. Intronic noncoding three base pair (TGA) deletion was present in splice site of HGF gene as a result of which protein features might be affected. Consequently, the genetic expression of c.482+1986_1988delTGA relates to the phenotypic manifestation of hereditary deafness in all four families.
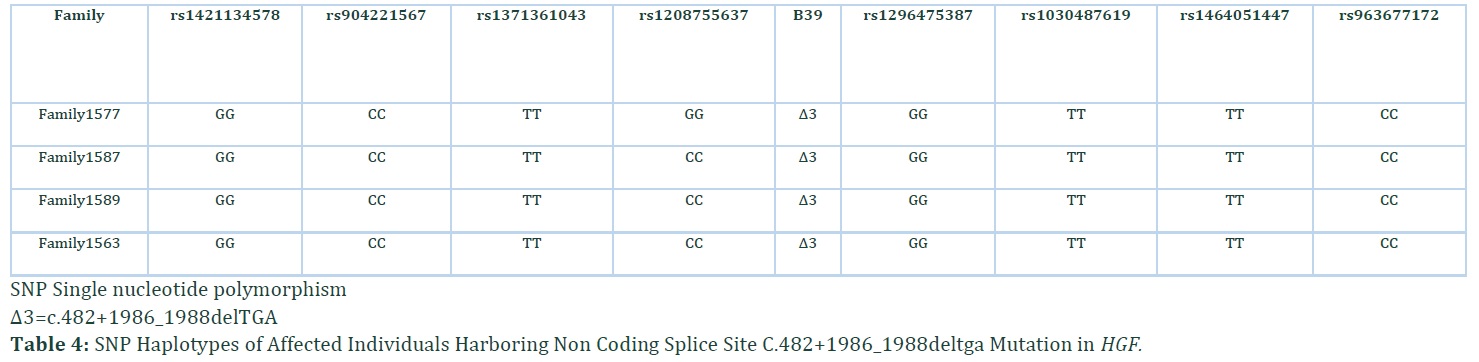
The damaging effects of this noncoding mutation were verified by using different in silico tools i.e., Mutation Taster, ExAC (exome aggregation consortium), LRT (likelihood ratio test), EVS (exome variant server), TGP (1000 genome project) and GERP (genomic evolutionary rate profiling) score (Table 3). The variant was classified as disease causing according to Mutation Taster. The occurrence of same causative pathogenic mutation (c.482+1986_1988delTGA) in all ethnically matched families inhabiting Kashmir region pave the mode to probe the founder effect of pathogenic noncoding mutation among these four Kashmiri families belonging to Jutt caste. All normal wild type bases flanking the detected mutation were assembled where established haplotype around site of mutation revealed the common founder effect of c.482+1986_1988delTGA mutation (Table 4).
Figures & Tables
Discussion![]()
In current study noncoding c.482+1986_1988delTGA HGF variant is reported in four consanguineous Kashmiri families with NSHD (non-syndromic hereditary deafness). The families were linked to DFNB39 locus at chromosome 7q21.11. Linkage was persistent to this region at distal (D7S660) and proximal side (D7S2540) with Zmax obtained with D7S2540 in all four families. The findings were consistent with the work of [15] ascertaining three base pair deletion in intron 4 (c.482+1986_1988delTGA) in thirty six Pakistani and two Indian families suffering from hearing loss. For this deletion mutation heterozygosity was detected in 2 out of 429 control samples of Pakistani population. While none of control sample out of 830 Indian, Caucasian, and human diversity panel control chromosomes revealed heterozygous genetic status for the same mutation. This noncoding deletion mutation vested in a region of putative splice enhancer sites (exonic) contiguous to an anticipated binding site of splice factor. The findings of present and previous study propose that the deletion might result in dysregulation of HGF isoforms [19].
All four families with same ethnicity and caste linked to DFNB39 locus presented same alleles haplotype strongly predicted this variant sharing common ancestral inheritance. Inconstant incidence of recurring variations in genes has been studied among ethnically diverse groups [20]. Three mutations (non-coding) in HGF have been reported in Pakistani and Indian population resulting in non-syndromic deafness. The two non-coding HGF variants in intron 4 were c.482+1986_1988delTGA and c.482+1991_2000delGATGATGAAA. The 3rd was c.495G>A; p.S165S synonymous splice site variant present in exon 5 [15]. In the present study, four out of forty five families suffering from hearing loss had non- coding deletion mutation, c.482+1986_1988delTGA, estimating 8.8% frequency of HGF variation in Kashmiri population which is in line with the previous study describing same frequency of HGF variation in a study on Pakistani families [21].
Our study encapsulates the genetic mutational spectrum concluding founder effect of detected HGF non-coding variation among hearing impaired families from Azad Jammu and Kashmir region of Pakistan. Increasing the screening of such variants will lessen disease burden and will further sweep the way of diagnostic therapeutics. This study outlines the mutational spectrum of HGF in Kashmiri population first time.
Abbreviations
HI: Hearing impairment
NSHD: Non syndromic hereditary deafness
HGF: Hepatocyte growth factor
ExAC: Exome aggregation consortium
LRT: Likelihood ratio test
EVS: Exome variant server
TGP: 1000 genome project
GERP: Genomic evolutionary rate profiling
Ethics approval and consent to participate
Ethical approval was obtained from the institutional review board, National Centre of Excellence in Molecular Biology, University of the Punjab, Lahore, Pakistan. After explanation of the objectives of this project, written informed consent was obtained from all participating individuals.
Consent for publication
Written informed consent to publish findings of this study was acquired from each participant.
Availability of data and material
All the data generated or analyzed during this study have been included in this manuscript.
Competing interests
The authors declare that they have no conflict of interest.
Funding
This study was supported by Higher Education Commission (HEC), Islamabad Pakistan. The funding agency had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authors' Contribution
KZ: study concept and design; identification and enrolment of families, implementation of experiments; data acquisition; analysis and interpretation of data; drafting of the manuscript. HT: analysis and interpretation of data; revising the manuscript critically for important intellectual content. SAM performed some lab tasks: AAK: supervised the work. All authors read and approved the final manuscript.
Acknowledgements
We are highly thankful to Higher Education Commission (HEC) of Pakistan for funding this work. Special thanks to Dr. S. Riazuddin, Dr. Shaheen N. Khan and Director CEMB, who guided and supported this study. The authors are grateful to Mr. M. Naeem and Mr. Asif for their help in collecting families, Dr. Asif Naeem, Ms. Hira Iqbal, Dr. Bushra Rauf and Bushra Irum for guiding in research tasks of this work and thank to all the affected and control individuals for their cooperation in this study.
The authors declare that they have no competing interests.
References![]()
- Elahi MM, Elahi F, Elahi A, Elahi SB. Paediatric hearing loss in rural Pakistan. Journal of Otolaryngology, (1998); 27(6): 348-353.
- Morton CC, Nance WE. Newborn hearing screening–a silent revolution. The New England Journal of Medicine, (2006); 354(20): 2151-2164.
- Shrivastava SR, Shrivastava PS, Ramasamy J. Supporting the global initiative of preventing childhood hearing loss: Act now, here's how! Noise Health, (2016); 18(84): 280-281.
- Ali A, Babar ME, Kalsoom S, Ahmad J, Abbas K. Linkage study of DFNB3 responsible for hearing loss in human. Indian Journal of Human Genetics (2013); 19(3): 325-330.
- Hussain R, Bittles AH. The prevalence and demographic characteristics of consanguineous marriages in Pakistan. Journal of Biosocial Science, (1998); 30(2): 261-275.
- Shami SA, Schmitt LH, Bittles AH. Consanguinity, spousal age at marriage and fertility in seven Pakistani Punjab cities. Annals of Human Biology, (1990); 17(2): 97-105.
- Meena R, Ayub M. Genetics Of Human Hereditary Hearing Impairment. Journal of Ayub Medical College Abbottabad, (2017); 29(4): 671-676.
- Friedman TB, Griffith AJ. Human nonsyndromic sensorineural deafness. Annual Review of Genomics and Human Genetics, (2003); 4341-402.
- Petersen MB, Wang Q, Willems PJ. Sex-linked deafness. Clinical Genetics, (2008); 73(1): 14-23.
- Petersen MB, Willems PJ. Non-syndromic, autosomal-recessive deafness. Clinical Genetics, (2006); 69(5): 371-392.
- Petit C, Levilliers J, Hardelin JP. Molecular genetics of hearing loss. Annual Review of Genetics, (2001); 35589-646.
- Schrijver I, Gardner P. Hereditary sensorineural hearing loss: advances in molecular genetics and mutation analysis. Expert Review of Molecular Diagnostics, (2006); 6(3): 375-386.
- van Wijk E, Krieger E, Kemperman MH, De Leenheer EM, Huygen PL, et al. A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26). Journal of Medical Genetics (2003); 40(12): 879-884.
- Zhu M, Yang T, Wei S, DeWan AT, Morell RJ, et al. Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). American Journal of Human Genetics, (2003); 73(5): 1082-1091.
- Schultz JM, Khan SN, Ahmed ZM, Riazuddin S, Waryah AM, et al. Noncoding mutations of HGF are associated with nonsyndromic hearing loss, DFNB39. American Journal of Human Genetics, (2009); 85(1): 25-39.
- Naz S, Friedman TB. Growth factor and receptor malfunctions associated with human genetic deafness. Clinical Genetics, (2020); 97(1): 138-155.
- Sambrook J, Russell DW. Purification of nucleic acids by extraction with phenol:chloroform. (2006); (1): 282-285. Cold Spring Harbor Protocols.
- Schaffer AA. Faster linkage analysis computations for pedigrees with loops or unused alleles. Human Heredity, (1996); 46(4): 226-235.
- Zhang YW, Vande Woude GF. HGF/SF-met signaling in the control of branching morphogenesis and invasion. Journal of Cellular Biochemistry, (2003); 88(2): 408-417.
- Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, et al. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. Journal of Medical Genetics (2003); 40(4): 242-248.
- Richard EM, Santos-Cortez RLP, Faridi R, Rehman AU, Lee K, et al. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Human Mutation, (2019); 40(1): 53-72.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0


