

Review Article
Personalized Medicine; a Potential Therapy for Cystic Fibrosis
Aqsa Ashraf1, Muhammad Usman Ghani2*, Muhammad Umer Khan3, Hafiz Muzzammel Rehman4, Mahmood ul Hassan1, Zohair Mehdi2
Adv. life sci., vol. 9, no. 4, pp. 437-445, December 2022
*– Corresponding Author: Muhammad Usman Ghani (Email: usman.camb@pu.edu.pk)
Authors' Affiliations
2. Centre for Applied Molecular Biology, University of the Punjab, Lahore- Pakistan.
3. Institute of Medical Lab Technology, Faculty of Allied Health Sciences, The University of Lahore- Pakistan
4. School of Biochemistry and Biotechnology, University of the Punjab, Lahore- Pakistan
[Date Received: 15/08/2022: Date Revised: 10/10/2022; Date Published: 31/12/2022]
Abstract![]()
Introduction
Methods
Discussion
Conclusion
References
Abstract
Cystic Fibrosis (CF) is an inherited disorder caused by mutations in CFTR gene that codes for Cystic Fibrosis Transmembrane-conductance Receptor anion channel. It is an autosomal recessive disease which affects the cells that secrete sweat, mucous and digestive juice, making these fluids thick and sticky, thus plugging ducts and tubes of various organs. The CF mutations are classified into various classes (class I, II, III, IV, V and VI) based on the cellular phenotype and complexity of mutants. The knowledge and understanding of biology and mechanisms of defects that underlie Cystic fibrosis paved a way to the development of different therapeutic approaches for these mutation classes. Ivacaftor first CFTR potentiator (FDA approved in 2012) is mostly used for Class III and IV mutations. Trials in patients with homozygous F508del mutation, a most common type of CF mutation that involves protein processing defects, showed no improvement with Ivacaftor alone, therefore, a double-combination therapy involving potentiator-corrector i.e., Ivacaftor-Lumacaftor got approval in 2015 to treat patients homozygous for F508del mutation. Then Ivacaftor-Tezacaftor (corrector) combination therapy was approved in 2018 which showed improved tolerability as compared to lumacaftor. In 2019, Trikfta, a triple combination therapy, came into light. It increases CFTR activity and is substantially considered to work more effectively in patients homozygous for F508del mutation. Studies and clinical trials reveal the outperformance of Trikafta in other available therapies in terms of respiratory symptoms, lungs functionality and quality of life on a whole.
Keywords: Cystic Fibrosis (CF); Cystic Fibrosis Transmembrane Conductance regulator (CFTR); Ivacaftor; Lumacaftor; Tezacaftor; Trikafta
Introduction![]()
Cystic Fibrosis (CF) is a severe form of autosomal recessive disease indicated by the aberrant transport of Epithelial electrolytes and results in elevation in the concentrations of sweat chloride, recurring lung infections, abnormal transport of bicarbonates and chlorides in pancreas and airways, and pancreatic insufficiency that ultimately leads to respiratory failure [1]. For centuries, high concentration of salts in sweat was the cardinal symptom of Cystic Fibrosis and was associated with mortality of infants. In 1930s, Dorothy Hansine noted the congested airways and pancreatic lesions in children who died due to coeliac disease and thus, comprehensively described Cystic Fibrosis for the first time [2]. In early 1980s, Dr. Paul Quinton noted that elevation in sweat electrolytes concentration in CF patients was due to the absence of the pathway for chloride reabsorption in CF sweat ducts [3]. In the same decade, efforts were made to identify the genetic basis of Cystic Fibrosis, which lead to the detection of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene in 1989 [4].
Cystic fibrosis (CF) is a genetic defect that has influence on different organs of about 70,000 to 100,000 people throughout the world [5]. It is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which affects the function and expression of CFTR protein, leading to defective transportation of chlorides and sodium ions across membrane of different organs, such as, pancreas, lungs, reproductive system, intestine and gall bladder [6].
Methods![]()
Literature Search and Selection Criteria
The literature was searched through Google scholar, Google Website, ScienceDirect, NCBI and FDA(USA) databases with the key words like “Cystic Fibrosis”, “CFTR”, “Cystic fibrosis therapy”, “Cystic Fibrosis Transmembrane Conductance regulator”, “Ivacaftor”, “Lumacaftor”, “Tezacaftor”, Trikafta and “Personalized medicine/drugs for Cystic Fibrosis’. Most recent articles were preferred for content referencing.
Molecular analysis of CFTR
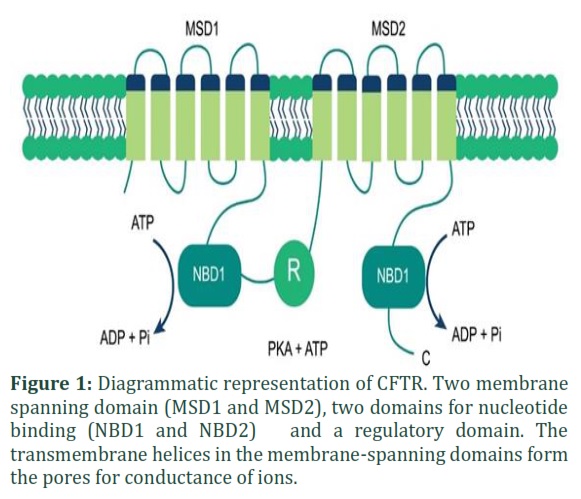
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) is a transmembrane channel for the bicarbonates and chloride ions. The CFTR gene is present at position q31-32 of long arm of chromosome 7 and encodes CFTR protein [7]. It has 1480 amino acids and is expressed in the membranes of the epithelial cells of some exocrine organs, and functions to regulate the fluid and salt homeostasis [8]. There are many membrane-integrated subunits of CFTR Glycoprotein that form two domains for nucleotide binding (NBD), two membrane-spanning domains (MSD1 and MSD2) and a regulatory domain (R) [9]. Membrane-spanning domain 1 (MSD1) and membrane-spanning domain 2 (MSD2) play their role in making channel pore walls and changes in their conformation induce the opening and closing of channel [10]. cAMP-dependent protein kinase A and C drive the phosphorylation of regulatory domain (R), which in turn enhances the association of nucleotide-binding domain (NBD) and ATP, mediates changes in their conformation and dimerize them in a head-to-tail conformation as shown in Figure 1. This conformation shows the channel’s opened state [11]. On the other hand, when ATP is hydrolyzed, it sets the channel to closed state [12].
The maturation of CFTR involves the complex folding of domain, assembly and MSD2 double N-glycosylation. During translation, process of CFTR maturation initiates in the Endoplasmic Reticulum and continue in Golgi Apparatus. In some instances, this process might mismatch and retard at various steps. Autophagy or ER-associated ubiquitin-dependent degradation system [13] then degrade this defected CFTR or sometimes even wild type CFTR protein. Upon reaching plasma membrane, clathrin-dependent endocytosis internalizes the wild type CFTR (WT- CFTR) and recycling endosomes recycle them back to surface of the cell [14].
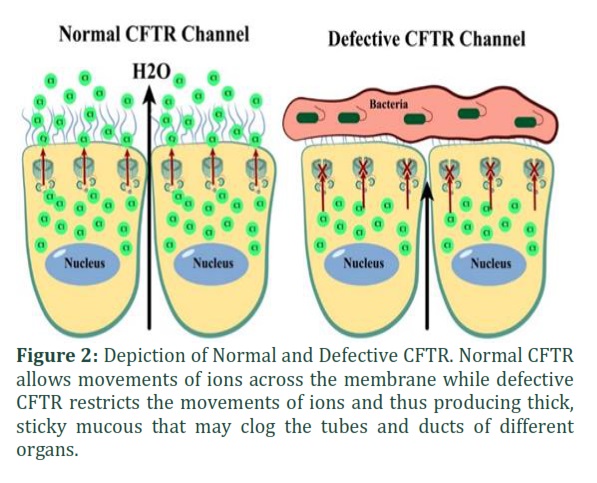
Cystic Fibrosis has been detected with more than 2000 mutations since the discovery of CFTR and has a direct influence of socioeconomic status of patients and the environmental conditions [15,16]. Lack in the production of functional CFTR causes defective secretion of bicarbonates (HCO3) and chloride (Cl-), coupled with more secretion of mucous and enhanced uptake of Na+. It dehydrates and acidifies the airway surface liquid [17]. The failed mucociliary clearance aggravates the inflammation and infections of lungs by bacteria (as shown in Figure 2) such as Aspergillus spp. and Pseudomonas aeruginosa, resulting in higher mortality and morbidity rates in CF patients [18].
Multi-Defects Etiologies
Pancreas
Apparently, there is a drastic impact of lack of CFTR on the exocrine pancreatic secretions. Damage of pancreas results in the inflammation, secretions of protein rich viscous fluids that obstructs the ducts by forming muco-protein plugs, destroy acini and form cysts in pancreas. The changes start in utero and lead to pancreatic insufficiency, which is apparent in 83% of Cystic Fibrosis patients. Common clinical manifestations of pancreatic insufficiency involve maldigestion, abdominal pain and decreased BMI [19], deficiency of fat-soluble vitamins, increased content of fats in feces that produce foul smelling and oily stool. Progression in the damage of pancreatic tissues lead to CF-related diabetes mellitus [20].
Gastrointestinal tract
Gastrointestinal tract is one of the earliest parts of the body that gets affected in Cystic Fibrosis. The exocrine pancreatic insufficiency alters the intraluminal milieu due to loss of pancreatic fluid rich in bicarbonates. The prolonged postprandial acidity due to less secretion of bicarbonates from lining of epithelial cells and the dehydration of lumen of intestine affects gastrointestinal tract. The acidic environment has drastic effects on the lining of cells and may also affects the function of digestive enzymes. The dehydration of intestine leads to intestinal obstruction due to build-up of viscous mucous. In neonatal patients of Cystic Fibrosis, this obstruction might result in meconium ileus and in adults, it might result in distal intestinal obstructive syndrome [20]. Thus, the loss of bicarbonates, a drop in the activity of digestive enzymes and decreased hydration of intestinal contents results in the malfunctioning of gastrointestinal tract [21].
Liver
The biliary epithelium of apical membrane expresses CFTR where it works in regulating the fluidity and alkalinity of bile. A defective CFTR may impair the alkalinity, hydration and secretion of the bile [22]. Progressive liver disease is regarded to be the third most prevalent cause of death in Cystic Fibrosis patients. Dehydrated and abnormally acidic bile is produced because of malfunctioned CFTR and clogs the bile ducts. This blockage of bile ducts activates pro-inflammatory response which leads to portal hypertension, multi-lobular cirrhosis, and fibrosis. Portal hypertension and cirrhosis increase the risk of early mortality [23].
Lungs
CFTR is highly expressed in the airway tissues. Non-functional or malfunctional CFTR causes decrease in the secretion of bicarbonates and cAMP-dependent chlorides into the airway secretions. In consequences, mucins bind to the apical surface of bronchi and the pH of airway surface fluid is decreased. Studies suggest that acidification of airway surface might induce the defects in the anti-bacterial defense of hosts [17]. Abnormal secretion of bicarbonates and chlorides tether mucus to the gland ducts [24]. Infections due to pathogens like hemophilus influenza, pseudomonas aeruginosa, klebsiella spp., and staphylococcus aureus lead to tissue destruction, systemic inflammation and respiratory insufficiency [25].
CFTR mutations and protein defects
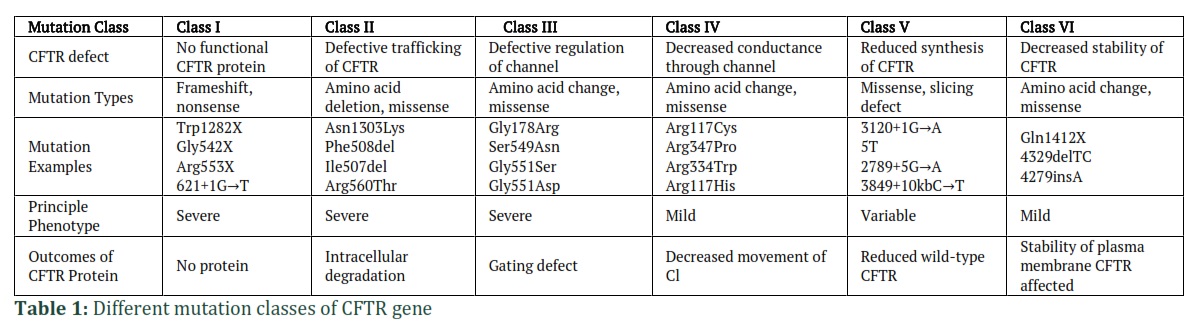
Since Cystic Fibrosis is an autosomal recessive genetic disorder, for CFTR gene to be affected, both copies of CFTR gene must have mutation. Mutations identified till to date have been classified into different classes (as illustrated in Table 1) on the basis of mechanism [20].
Class I mutations
In class I mutations, there is reduction in the production of CFTR protein or it may be absent completely or partially. This loss of CFTR protein arise either from non-sense mutation (premature termination codon), frameshift mutation (deletion or insertion/ indel) or splicing defects of mRNA [26]. Mutations in W1282X and G542X generate premature termination codons (PTC) and produce truncated proteins. Loss in the levels of CFTR mRNA are also observed in nonsense-mediated decay in which mRNA species containing abnormal PTC are degraded [27]. The most common mutations of this class are Trp1282X, Gly542X and Arg553X, counting up to 7% patients of Cystic Fibrosis [20].
Class II mutations
Class II mutations associate with the inappropriate processing of protein due to poor stability or misfolding of protein. The resulting protein is retained in the Endoplasmic Reticulum where it is degraded by the proteasomes [28]. This mutation results in defects in the post-translational modification of CFTR protein. Common mutation examples of this class are Asn1303Lys, Phe508del, Ile507del, Arg560Thr. Class II mutation prevail in up to 85% of Cystic Fibrosis patients because of F508del mutation. F508del mutation is the most prevalent type of mutation. In 45% of Cystic Fibrosis patients, this mutation is present on both alleles, while 70% patients have this mutation on one allele. [20].
Class III mutations
Class III mutations are the missense mutations which also termed as gating mutations and are present in the ATP-binding domain of CFTR protein. These mutations have little to no impact on how proteins are processed. The mutant proteins get inserted to plasma membrane but do not activate or gate normally due to phosphorylation of protein kinase. The prominent mutation of this class is G551D [29] and other mutations include Gly178Arg, Ser549Asn. [30].
Class IV mutations
This class of mutations is present within the pore-forming region of membrane spanning domain. This missense mutation produces a protein and export it to surface of cell from Endoplasmic Reticulum. Phosphorylation of protein kinase activates the channel, but this channel has little capacity for conducting the anions through its pores. Class IV mutation is a milder form since the ability of channel to conduct anions is lowered, rather than a complete abolishment of conductance. R117H mutation is the best suited in this class, forming a properly processed protein and a chloride channel which has reduced conductance [31]. Some other examples include Arg117Cys, Arg347Pro, Arg334Trp mutations [30].
Class V mutations
In class V mutations, the limitation in the transcriptional regulation reduces the production of CFTR protein [32]. Splicing error is produced which affects the CFTR mRNA stability and ultimately decreases the expression levels of CFTR. Common examples of class V mutations include c.1680-886A˃G, A455E and c.2657+5G˃A and accounts for less than 3% cases of CF [20].
Class VI mutation
In class VI mutations, the stability of CFTR is affected and results in the increased turnover of CFTR protein [33]. This mutation class destabilizes the CFTR channel in plasma membrane or in post-ER compartments by reducing the conformational stability of channel [34] and thus resulting in increased turnover rate of protein [35]. Examples of class VI mutations include Gln1412X, 4329delTC, 4279insA [30].
CF Diagnosis
Most of Cystic Fibrosis cases are diagnosed through newborn screening strategy [36]. Screening of newborns has a lot of benefits like early diagnosis, malnutrition prevention, slowing down the progression of lung disease and providing medical and psychosocial support to the victims of CF and their families [37]. The newborn screening is performed using Immunoreactive Trypsinogen: IRT/DNA and IRT/IRT1/DNA [38]. Immunoreactive Trypsinogen (IRT) is protein marker to check for the pancreatic diseases and inflammation and is regarded as the initial indicator of positive screening results [36]. Specificity and sensitivity of screening is improved by employing DNA testing in combination with IRT [39]. The IRT/DNA strategy identifies the mutation in CFTR gene. It identifies patients that have one or two copies of mutated genes. If both copies are mutated, the patient is diagnosed with disease, but if one mutated copy is present the patients are identified as carriers of CF.
When patient has Immunoreactive Trypsinogen level greater than 60ng/mL, then IRT/IRT1/DNA plays clinically significant role, and the test is repeated within two weeks. CFTR mutation analysis is done on patient’s DNA if IRT levels are high. This method has sensitivity levels of more than 99.5% [40]. After an initial screening of newborns by IRT/DNA or IRT/IRT1/DNA, patients undergo sweat chloride test. It measures quantitatively the concentration of chlorides present in sweat and if concentration exceeds 60mmol/L, CF is diagnosed [41].
Existing personalized therapies
Since discovery of Cystic Fibrosis gene in 1989, researchers have been attempting to figure out how mutations in CFTR gene manifest functional and biochemical abnormalities in its protein. Knowledge of these dysfunctionalities paved a way to the production of pharmacologic agents for targeting these anomalies. Correctors and potentiators of CFTR became available to treat patients of Cystic Fibrosis (Sala MA et al., 2018). FDA have approved these two classes of CFTR modulators (correctors/potentiators) to treat CF patients.
Ivacaftor
Potentiators work efficiently against class III and IV mutations [42]. These potentiators bind with CFTR protein and increase the frequency of channel opening and conductance of ions. The FDA approved potentiator of CFTR is Ivacaftor (IVA). The collaborative research between Vertex Pharmaceuticals and Cystic Fibrosis Foundation initiated a high throughput screening (HTS) utilizing a library of 228,000 compounds, which led to discovery of Ivacaftor potentiator (VX-770, trade name Kalydeco) [29]. It was originally licensed by FDA for CF patients with G551D mutation. The primary target of Ivacaftor is CFTR protein in which Glycine is switched by Aspartic acid at position 551 (thus named G551D). Patients with multiple CFTR gating mutations are recommended to get Ivacaftor treatment [43]. The exact molecular mechanism of Ivacaftor for increasing the opening probability of CFTR channel is not clear but work done by Hwang and colleagues reveal that Ivacaftor stabilize the post-hydrolytic open state of channel which leads to uncoupling of ATP hydrolysis and increase the open state residence time [44]. Even though G551D is the second most prevalent mutation of CFTR, it is present only in 4-5% of CF patients and thus Ivacaftor alone is not of a benefit for many patients. Though it was initially approved to treat G551D mutation patients, it is considered to work well against some other gating mutations like G1349D, S1255P, G970R, S1251N, G1244E, G551S, S549R, and G178R [45]. Two random placebo-controlled trials were made to evaluate the safety and effectiveness of Ivacaftor in patients that had at least one G551D mutation in CFTR. Patients who received this drug were observed to undergo changes in their CF manifestations and had a risk of pulmonary exacerbation that was 55% lower than that of patients who received placebo [46]. Ivacaftor significantly reduced concentration of sweat chlorides but could not enhance function of lungs, indicating that a potentiator alone was insufficient for this mutant protein [47]. Studies also revealed an increase in activity of innate immune cell by Ivacaftor [48].
Ivacaftor-Lumacaftor
In 2015, a combination therapy involving Lumacaftor (Corrector) and Ivacaftor (Potentiator- Orkambi) was approved by FDA, to be used in patients (over 12 years of age) who are homozygous for F508del mutation [49]. Since Ivacaftor assists in increasing the conduction of chlorides through cell epithelium and increase the time for opened state of channels, and Lumacaftor helps in the delivery of F508del-CFTR protein to surface of cell, these drugs, when combined, show improvement in the underlying defect of F508del [50]. The in vitro studies reveal 15% increase in the transport of CFTR chloride by Lumacaftor alone, and a 30% increase by combination of Lumacaftor and Ivacaftor. As observed in F508del mutation, the conductance defect and abnormal folding of CFTR can therefore be addressed by combining a corrector and a potentiator [51].
Ivacaftor-Tezacaftor
Tezacaftor is a new CFTR corrector recently approved by FDA to be used in conjugation with Ivacaftor. This combination therapy was found to be effective in patients with homozygous F508del mutation and residual function mutation of the second allele. It shows 2.5 to 8.1% enhancement in trafficking and processing of F508del-CFTR and in transport of chloride in bronchial epithelial cells of human as compared to normal cells. This combination is considered more effective than Ivacaftor-Lumacaftor in terms of FEV1 improvements as well as its adverse effects are also less than Ivacaftor-Lumacaftor [52].
Elexacaftor
Elexacaftor is a next-generation corrector of CFTR because its mechanism and structure differ from those of correctors like Tezacaftor. It is a better therapy for those patients who have heterozygous F508del-CFTR mutation and for genes that produce proteins which are resistant to Tezacaftor or Ivacaftor [53]. This CFTR corrector modulates the CFTR protein so that it boosts the trafficking of mature CFTR proteins on the cell’s surface. If this drug is used in combination with a CFTR potentiator, it might improve multi-organ symptoms of cystic fibrosis, including nutritional status, functioning of lungs and quality of life altogether [53].
Trikafta
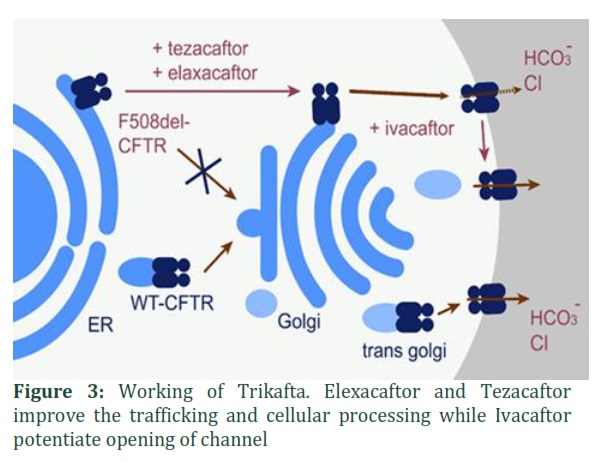
In 2019, FDA approved a triple combination therapy, Trikafta, which is a combination of one potentiator (Ivacaftor) and two correctors (Tezacaftor and Elexacaftor) (illustrated in Figure 3). Ivacaftor is thought to directly work on mutant channel forming protein of CFTR. Elexacaftor and Tezacaftor make a directly interact with a mutant F508del-CFTR polypeptide. But the main target of Trikafta is the epithelial cell linings of all the tubular systems that get afflicted by Cystic Fibrosis, i.e., pancreas, lungs, liver and gastrointestinal tract [54]. Two double-blind, random phase three studies were conducted in patients who were 12 years or more in age and had at least one F508del mutation, after that, FDA approved this drug. In first trial, 400 patients were involved, and it was a placebo-controlled trial of 24-weeks. Second trial lasted for four weeks and involved 100 patients having two comparable F508del mutations. In these trials, pharmacokinetics, tolerability, acceptability, performance and safety of the drug were tested and studies revealed improvement in respiratory symptoms, sweat chloride, pulmonary exacerbation and lung functions [55]. Since FEV1 (forced expiratory volume) and pulmonary exacerbation are important predictors of clinical status of CF patients, lowering of pulmonary exacerbations and improvement in FEV1 was observed in these trials [56].
Portraiture on a wider frame; some Real-world experiences
Although the modulator therapy has taken the world of research as a storm and has a great potential, still the fact cannot be neglected that there is a cost associated with personalized medicine. The clinical trials for the afore mentioned drugs enabled us to have access on modulator therapies. However, these trials were made on a specific population following strict exclusion and inclusion criteria. Data extracted from the “real-world” after these observational studies would better expand our knowledge on the facts. Being the first licensed modulator of CFTR, majority of the real-world data relates to the working and effectiveness of Ivacaftor in treatments [57]. Duckers and team reviewed the impacts of Ivacaftor on pediatric population and in adults for up to a period of 5 years and revealed that majority of cases showed an increment of 5-10 percentage in the mean absolute change in ppFEV1 from baseline [58]. A study performed on adults with G551D revealed the benefits were not sustained in ppFEV1 and ppFEV1 declined to pre-treatment levels in 5 years [59]. Studies also showed that benefits in ppFEV1 were sustained more in adults than in children [60]. Significant decrease in pulmonary exacerbations and infectious attacks of Aspergillus spp. and Pseudomonas aeruginosa were also decreased. Quality of life was reported to be improved and maintained in 5.5years [60].
Impacts of Lumacaftor/Ivacaftor treatment were studied in French multi-center study and it was demonstrated that treatment was correlated with improvements in ppFEV1. Adverse effects in respiration including bronchospasm and tightness of chest, were observed in patients with advanced disease and thus eighteen percent of patients discontinued their treatment [61].
Real-world impacts of Elexacaftor/Tezacaftor/Ivacaftor triple therapy are not much prominent yet due to its recent approval but there are some indications of improvements in patients with severe disease of lungs, yet there is a need for more efficient treatments to appear [62].
Post-licensing studies revealed more adverse events related to Lumacaftor/Ivacaftor usage as compared to other modulators. Respiratory and lung diseases are observed, and their symptoms include tightening of chest and dyspnea. These symptoms often prevail when treatment is initiated and later discontinues in up to 30% of patients [63]. These symptoms do not relate to use of other modulator combinations or even to use of Ivacaftor alone, and so it is thought to be specific to Lumacaftor. Clinical trials of Lumacaftor/Ivacaftor and Elexacaftor/Tezacaftor expressed elevation in blood pressure. Fatigue, headache and derangement of liver enzymes are reported to be related to Lumacaftor/Ivacaftor and rash is linked with usage of all types of modulators [63]. Elevation in the levels of creatinine kinase is also reported to be a rare event related to all modulators.
Figures & Tables
Amplifiers
Amplifiers are the compounds that increase the amount of messenger RNA of mutant CFTR, resulting in increased amount of CFTR protein and thus the substrates of other modulators of CFTR. High throughput screening was of help to identify these amplifiers. They relate to variants of class V mutation or can be used in combination with potentiators and correctors or can be used with all variants, other than those that do not produce mRNA. Their effects showed benefits in preclinical studies but still are to be investigated in clinical trials [57].
Read-Through agents
Despite many successful CFTR modulators have been developed to treat Cystic Fibrosis, still other classes of CFTR variants are needed to be studied. premature termination codons (PTCs) are the cause of class I mutation and result in defective CFTR protein [64]. Such variants may be treated by read-through agents that result in production of full-length CFTR. The read-through agents of PTC mutation replace the mutant stop codon with another amino acid. Insertion of amino acid is random, but preference is mostly given to tyrosine, tryptophan, glutamine, arginine or cytosine [65].Ataluren is a read-through agent that showed promising results when discovered but it is not of much benefit in larger clinical trials [66]. Some other read-through agents like ELX-02 are going through clinical investigations. In preclinical studies, ELX-02 showed a dose-dependent read-through of nonsense mutation to yield full-length CFTR protein, but the compound is still under study [67].
Some other modulators
Some CFTR modulators are on their way to development. These include corrector Navocaftor (ABBV-3067) and potentiator Galicaftor (ABBV-2222). ABBV-2222 was checked for its working in lung functions in the phase 2 clinical studies, but it did not show significant improvements alone. In combination with ABBV-3067, ABBV-2222 was found well tolerated and is under investigation [68]. Another potentiator, ABBV-119 is under clinical trials for its triple combination therapy with Galicaftor and Navocaftor [57].
Until few years back, the DNA mutation analysis of CF patients was less a clinical issue and more a research issue. However, after the discovery of CFTR modulators, whole frame is changed. Now a detailed knowledge of patient’s CFTR mutation is requisite for determining appropriate treatment strategy and whether a potentiator or a corrector would be of benefit to patient. In a shorter period, the CF treatment approaches evolved from one-size-fits-all to a wide treatment variety, based on mutations in CFTR gene of a patient. In nutshell, genomics and molecular genetics have paved the way to an Era of Personalized Medicine.
Author Contributions
Aqsa Ashraf: Manuscript writing and literature survey.
Muhammad Usman Ghani: Conceived the idea, literature survey, manuscript editing and drafting.
Muhammad Umer Khan: Literature survey, proofreading and editing of manuscript.
Hafiz Muzzammel Rehman: Manuscript writing, literature survey.
Mahmood ul Hassan: Designee and interpretation of data.
Zohair Mehdi: Manuscript writing and editing.
The authors have declare that there is no conflict of interest.![]()
References
- Bernardino AL, Lima CE, Zatz M. Analysis of mutations in the cystic fibrosis transmembrane regulator (CFTR) gene in patients with obstructive azoospermia. Genetics and molecular biology, (2003); 261-3.
- Bradbury NA (2020) CFTR and Cystic Fibrosis: A Need for Personalized Medicine. In: Hamilton KL, Devor DC, editors. Studies of Epithelial Transporters and Ion Channels Physiology in Health and Disease. Cham: Springer. pp. 773-802.
- Quinton PM. Chloride impermeability in cystic fibrosis. Nature, (1983); 301(5899): 421-422.
- Riordan JR, Rommens JM, Kerem B-s, Alon N, Rozmahel R, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science, (1989); 245(4922): 1066-1073.
- Kelly J. Environmental scan of cystic fibrosis research worldwide. Journal of Cystic Fibrosis, (2017); 16(3): 367-370.
- Flume PA, Robinson KA, O'Sullivan BP, Finder JD, Vender RL, et al. Cystic fibrosis pulmonary guidelines: airway clearance therapies. Respiratory care, (2009); 54(4): 522-537.
- Alibakhshi R, Mohammadi A, Khamooshian S, Kazeminia M, Moradi K. CFTR gene mutation spectrum among 735 Iranian patients with cystic fibrosis: A comprehensive systematic review. Pediatric pulmonology, (2021); 56(12): 3644-3656.
- Linsdell P. Functional architecture of the CFTR chloride channel. Molecular membrane biology, (2014); 31(1): 1-16.
- Rowe S. Miller S, Sorscher EJ. Cystic fibrosis N Engl J Med, (2005); 3521992-2001.
- Muallem D, Vergani P. ATP hydrolysis-driven gating in cystic fibrosis transmembrane conductance regulator. Philosophical Transactions of the Royal Society B: Biological Sciences, (2009); 364(1514): 247-255.
- Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem, (2008); 77701-726.
- Vergani P. Lockless SW, Nairn AC, Gadsby DC. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains Nature, (2005); 433876-880.
- Gelman MS, Kannegaard ES, Kopito RR. A principal role for the proteasome in endoplasmic reticulum-associated degradation of misfolded intracellular cystic fibrosis transmembrane conductance regulator. Journal of Biological Chemistry, (2002); 277(14): 11709-11714.
- Sharma M, Pampinella F, Nemes C, Benharouga M, So J, et al. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. The Journal of cell biology, (2004); 164(6): 923-933.
- Kopp BT, Sarzynski L, Khalfoun S, Hayes Jr D, Thompson R, et al. Detrimental effects of secondhand smoke exposure on infants with cystic fibrosis. Pediatric Pulmonology, (2015); 50(1): 25-34.
- Ghani MU, Sabar MF, Awan FI. WS21.03 Low-cost chain termination DNA sequencing PCR reaction to diagnose CFTR gene mutations. Journal of Cystic Fibrosis, (2022); 21S41-S42.
- Pezzulo AA, Tang XX, Hoegger MJ, Abou Alaiwa MH, Ramachandran S, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature, (2012); 487(7405): 109-113.
- Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. New England Journal of Medicine, (2015); 372(4): 351-362.
- Madácsy T, Pallagi P, Maleth J. Cystic fibrosis of the pancreas: the role of CFTR channel in the regulation of intracellular Ca2+ signaling and mitochondrial function in the exocrine pancreas. Frontiers in Physiology, (2018); 1585.
- Harutyunyan M, Huang Y, Mun K-S, Yang F, Arora K, et al. Personalized medicine in CF: from modulator development to therapy for cystic fibrosis patients with rare CFTR mutations. American Journal of Physiology-Lung Cellular and Molecular Physiology, (2018); 314(4): L529-L543.
- Kopelman H, Durie P, Gaskin K, Weizman Z, Forstner G. Pancreatic fluid secretion and protein hyperconcentration in cystic fibrosis. New England Journal of Medicine, (1985); 312(6): 329-334.
- Strazzabosco M, Fabris L, Spirli C. Pathophysiology of cholangiopathies. Journal of clinical gastroenterology, (2005); 39(4): S90-S102.
- Ledder O, Haller W, Couper RT, Lewindon P, Oliver M. Cystic fibrosis: an update for clinicians. Part 2: hepatobiliary and pancreatic manifestations. Journal of gastroenterology and hepatology, (2014); 29(12): 1954-1962.
- Hoegger MJ, Fischer AJ, McMenimen JD, Ostedgaard LS, Tucker AJ, et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science, (2014); 345(6198): 818-822.
- Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med, (2007); 58157-170.
- Rowntree RK, Harris A. The phenotypic consequences of CFTR mutations. Annals of human genetics, (2003); 67(5): 471-485.
- Pranke I, Bidou L, Martin N, Blanchet S, Hatton A, et al. Factors influencing readthrough therapy for frequent cystic fibrosis premature termination codons. ERJ open research, (2018); 4(1).
- Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, et al. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell, (1995); 83(1): 129-135.
- Bradbury NA (2020) CFTR and cystic fibrosis: a need for personalized medicine. Studies of Epithelial Transporters and Ion Channels: Springer. pp. 547-604.
- Bell SC, Mall MA, Gutierrez H, Macek M, Madge S, et al. The future of cystic fibrosis care: a global perspective. The Lancet Respiratory Medicine, (2020); 8(1): 65-124.
- Reddy M, Quinton PM. Selective activation of cystic fibrosis transmembrane conductance regulator Cl-and HCO3-conductances. Jop, (2001); 2(4 Suppl): 212-218.
- Beck S, Penque D, Garcia S, Gomes A, Farinha C, et al. Cystic fibrosis patients with the 3272‐26A→ G mutation have mild disease, leaky alternative mRNA splicing, and CFTR protein at the cell membrane. Human mutation, (1999); 14(2): 133-144.
- Ramalho AS, Lewandowska MA, Farinha CM, Mendes F, Gonçalves J, et al. Deletion of CFTR translation start site reveals functional isoforms of the protein in CF patients. Cellular Physiology and Biochemistry, (2009); 24(5-6): 335-346.
- Haardt M, Benharouga M, Lechardeur D, Kartner N, Lukacs GL. C-terminal truncations destabilize the cystic fibrosis transmembrane conductance regulator without impairing its biogenesis: a novel class of mutation. Journal of Biological Chemistry, (1999); 274(31): 21873-21877.
- Silvis MR, Picciano JA, Bertrand C, Weixel K, Bridges RJ, et al. A mutation in the cystic fibrosis transmembrane conductance regulator generates a novel internalization sequence and enhances endocytic rates. Journal of Biological Chemistry, (2003); 278(13): 11554-11560.
- Shenoy A, Spyropoulos D, Peeke K, Smith D, Cellucci M, et al. Newborn Screening for Cystic Fibrosis: Infant and Laboratory Factors Affecting Successful Sweat Test Completion. International journal of neonatal screening, (2020); 7(1): 1.
- Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. The Journal of pediatrics, (2008); 153(2): S4-S14.
- Sontag MK, Lee R, Wright D, Freedenberg D, Sagel SD. Improving the sensitivity and positive predictive value in a cystic fibrosis newborn screening program using a repeat immunoreactive trypsinogen and genetic analysis. The Journal of Pediatrics, (2016); 175150-158. e151.
- Baker MW, Groose M, Hoffman G, Rock M, Levy H, et al. Optimal DNA tier for the IRT/DNA algorithm determined by CFTR mutation results over 14 years of newborn screening. Journal of Cystic Fibrosis, (2011); 10(4): 278-281.
- Sontag MK, Wright D, Beebe J, Accurso FJ, Sagel SD. A new cystic fibrosis newborn screening algorithm: IRT/IRT1↑/DNA. The Journal of pediatrics, (2009); 155(5): 618-622.
- Rock MJ, Makholm L, Eickhoff J. A new method of sweat testing: the CF Quantum® sweat test. Journal of Cystic Fibrosis, (2014); 13(5): 520-527.
- Rogan MP, Stoltz DA, Hornick DB. Cystic fibrosis transmembrane conductance regulator intracellular processing, trafficking, and opportunities for mutation-specific treatment. Chest, (2011); 139(6): 1480-1490.
- Lopes-Pacheco M. CFTR modulators: shedding light on precision medicine for cystic fibrosis. Frontiers in pharmacology, (2016); 7275.
- Jih K-Y, Hwang T-C. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proceedings of the National Academy of Sciences, (2013); 110(11): 4404-4409.
- Yu H, Burton B, Huang C-J, Worley J, Cao D, et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. Journal of Cystic Fibrosis, (2012); 11(3): 237-245.
- Davis PB, Yasothan U, Kirkpatrick P. Ivacaftor. Nature reviews Drug discovery, (2012); 11(5): 349-351.
- Deeks ED. Lumacaftor/ivacaftor: a review in cystic fibrosis. Drugs, (2016); 76(12): 1191-1201.
- Favia M, Gallo C, Guerra L, De Venuto D, Diana A, et al. Treatment of cystic fibrosis patients homozygous for F508del with lumacaftor-ivacaftor (Orkambi®) restores defective CFTR channel function in circulating mononuclear cells. International journal of molecular sciences, (2020); 21(7): 2398.
- Sala MA, Jain M. Tezacaftor for the treatment of cystic fibrosis. Expert review of respiratory medicine, (2018); 12(9): 725-732.
- Quintana-Gallego E, Delgado-Pecellín I, Acuña CC. CFTR protein repair therapy in cystic fibrosis. Archivos de Bronconeumología (English Edition), (2014); 50(4): 146-150.
- Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. The lancet Respiratory medicine, (2014); 2(7): 527-538.
- Gentzsch M, Mall MA. Ion channel modulators in cystic fibrosis. Chest, (2018); 154(2): 383-393.
- Taylor-Cousar JL, Mall MA, Ramsey BW, McKone EF, Tullis E, et al. Clinical development of triple-combination CFTR modulators for cystic fibrosis patients with one or two F508del alleles. ERJ open research, (2019); 5(2).
- Zaher A, ElSaygh J, Elsori D, ElSaygh H, Sanni A. A Review of Trikafta: Triple Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulator Therapy. Cureus, (2021); 13(7).
- Keating D, Marigowda G, Burr L, Daines C, Mall MA, et al. VX-445–tezacaftor–ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. New england journal of medicine, (2018); 379(17): 1612-1620.
- Konstan MW, Wagener JS, VanDevanter DR, Pasta DJ, Yegin A, et al. Risk factors for rate of decline in FEV1 in adults with cystic fibrosis. Journal of Cystic Fibrosis, (2012); 11(5): 405-411.
- Haq I, Almulhem M, Soars S, Poulton D, Brodlie M. Precision Medicine Based on CFTR Genotype for People with Cystic Fibrosis. Pharmacogenomics and Personalized Medicine, (2022); 1591.
- Duckers J, Lesher B, Thorat T, Lucas E, McGarry LJ, et al. Real-world outcomes of ivacaftor treatment in people with cystic fibrosis: a systematic review. Journal of clinical medicine, (2021); 10(7): 1527.
- Mitchell RM, Jones AM, Stocking K, Foden P, Barry PJ. Longitudinal effects of ivacaftor and medicine possession ratio in people with the Gly551Asp mutation: a 5-year study. Thorax, (2021); 76(9): 874-879.
- Guimbellot J, Baines A, Paynter A, Heltshe S, VanDalfsen J, et al. Long term clinical effectiveness of ivacaftor in people with the G551D CFTR mutation. Journal of Cystic Fibrosis, (2021); 20(2): 213-219.
- Burgel P-R, Munck A, Durieu I, Chiron R, Mely L, et al. Real-life safety and effectiveness of lumacaftor–ivacaftor in patients with cystic fibrosis. American journal of respiratory and critical care medicine, (2020); 201(2): 188-197.
- Benden C, Schwarz C. CFTR Modulator Therapy and Its Impact on Lung Transplantation in Cystic Fibrosis. Pulmonary Therapy, (2021); 7(2): 377-393.
- Dagenais RV, Su VC, Quon BS. Real-world safety of CFTR modulators in the treatment of cystic fibrosis: a systematic review. Journal of clinical medicine, (2020); 10(01): 23.
- Zainal Abidin N, Haq IJ, Gardner AI, Brodlie M. Ataluren in cystic fibrosis: development, clinical studies and where are we now? Expert opinion on pharmacotherapy, (2017); 18(13): 1363-1371.
- Blanchet S, Cornu D, Argentini M, Namy O. New insights into the incorporation of natural suppressor tRNAs at stop codons in Saccharomyces cerevisiae. Nucleic acids research, (2014); 42(15): 10061-10072.
- Konstan M, VanDevanter D, Rowe S, Wilschanski M, Kerem E, et al. Efficacy and safety of ataluren in patients with nonsense-mutation cystic fibrosis not receiving chronic inhaled aminoglycosides: the international, randomized, double-blind, placebo-controlled Ataluren Confirmatory Trial in Cystic Fibrosis (ACT CF). Journal of Cystic fibrosis, (2020); 19(4): 595-601.
- Kerem E. ELX-02: An investigational read-through agent for the treatment of nonsense mutation-related genetic disease. Expert Opinion on Investigational Drugs, (2020); 29(12): 1347-1354.
- Bell SC, Barry PJ, De Boeck K, Drevinek P, Elborn JS, et al. CFTR activity is enhanced by the novel corrector GLPG2222, given with and without ivacaftor in two randomized trials. Journal of Cystic Fibrosis, (2019); 18(5): 700-707.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0
![]()