

Full Length Research Article
Identification of potential inhibitors targeting DNA adenine methyltransferase of Klebsiella pneumoniae for antimicrobial resistance management: a structure-based molecular docking study
Ameerah Ali Alshehri1, Ayshah Musad Almutairi2, Alaa Shafie3, Norah Ali Alshehri4,5, Samia Musaad Almutairi6, Farah Anjum3*
Adv. life sci., vol. 10, no. 4, pp. 604-608, December 2023
*– Corresponding Author: Farah Anjum (farahanjum@tu.edu.sa)
Authors' Affiliations
2. Armed Forces Center for Health Rehabilitation, AlTaif – Saudi Arabia
3. Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, Taif University, Taif – Saudi Arabia
4. Department of Family and Community Medicine, College of Medicine, King Saud University (KSU), Riyadh – Saudi Arabia
5. University Family Medicine Center, King Saud University Medical City, King Saud University, Riyadh – Saudi Arabia
6. Taif Directorate of Health Affairs, AlTaif – Saudi Arabia
[Date Received: 17/07/2023; Date Revised: 17/08/2023; Date Published Online: 31/12/2023; Date Updated:09/09/2025]
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Klebsiella pneumoniae is an important opportunistic pathogen that frequently causes nosocomial infections. Notably, this bacterium has emerged as a significant problem in hospital settings because of its acquisition of resistance to carbapenems. The majority of antibiotics act by targeting crucial pathways within bacterial cells. However, due to the development of resistance mechanisms, the efficiency of these antibiotics has decreased. Therefore, this study focused on a putative protein (DNA adenine methyltransferase; Dam) found in K. pneumoniae that encompasses a DNA methylation protein domain, indicating a novel potential target for pharmacological intervention. DNA methylation affects bacterial virulence attenuation.
Methods: Due to the absence of an available 3D structure for the Dam protein in the protein database, a 3D model was generated using the SWISS-MODEL server and validated using computational tools. Following that, screening was performed against the Dam protein utilizing a set of 2706 phytochemicals obtained from the ZINC database using PyRx0.8. The ProTox-II platform was used to predict the physicochemical properties and various toxicity endpoints.
Results: Among the screened compounds, ZINC4214775, ZINC4095704, and ZINC4136964 had higher binding affinity for the Dam and interacted with its active site residues. The computational analyses of these three identified hits indicate that their predicted properties were within an acceptable range for evaluating toxicity. In addition, a toxicity radar chart showed that these hits were within an acceptable range.
Conclusions: These compounds have the potential to act as Dam inhibitors and could be investigated further for managing antimicrobial resistance in K. pneumoniae.
Keywords: Klebsiella pneumoniae; Dam; Antimicrobial resistance; Phytochemicals
Introduction![]()
Klebsiella pneumoniae, a member of the Enterobacteriaceae family, is an indigenous commensal member of the human and animal gastrointestinal microbiota [1]. This bacterium has multiple antibiotic resistance mechanisms and frequently acts as a pathogen, causing nosocomial infections such as surgical wound infections, gastrointestinal infections, and community-onset infections, and often causes outbreaks within healthcare facilities. The global prevalence of drug-resistant K. pneumoniae has increased significantly, reaching up to 70%, and the associated mortality rate from infections caused by this pathogen ranges between 40% to 70% [2]. The emergence of multidrug-resistant as well as carbapenem-resistant K. pneumoniae has become a significant global public health concern [3].
DNA adenine methyltransferase (Dam) is a promising target for therapeutic intervention because it plays an essential role in the epigenetic regulatory apparatus that is responsible for bacterial pathogen survival and pathogenicity [4]. Dam has been implicated in a variety of cellular processes such as mismatch repair [5], replication regulation, transposition [6], and gene expression control [7]. Furthermore, studies have found a link between Dams and antibiotic resistance. Elevated resistance to ampicillin, tetracycline, and nalidixic acid has been observed in bacteria due to epigenetically induced changes in the expression of resistance-associated genes [8].
The process of introducing a new pharmaceutical medication to the market necessitates a significant financial and time investment. The average time for drug development is 10-15 years, with a cost estimate of approximately $800 million [9,10]. Computer-aided drug discovery (CADD) has emerged as an essential component of numerous projects conducted in a variety of settings and research environments. CADD has significantly aided in the identification and optimization of hit compounds, propelling them to advanced stages of the drug discovery pipeline or eventual market entry [11]. This study aimed to find natural Dam inhibitors to fight the antimicrobial resistance in K. pneumoniae.
Methods![]()
Sequence retrieval and homology modeling of the Dam protein
The UniProt database was used to find K. pneumoniae protein sequences containing the Dam domain (UniProt ID: A0A2U0NNR3). The SWISS-MODEL server was used to build the targeted 3D protein structure, which involved fully automated procedures. The structure of the protein of interest was first generated using homology modeling, which was then compared to a suitable template protein structure [12]. The structural properties of the Dam-containing protein in E. coli (PDB ID: 4RTL) were used to select the template protein [13].
Validation of the modeled structure
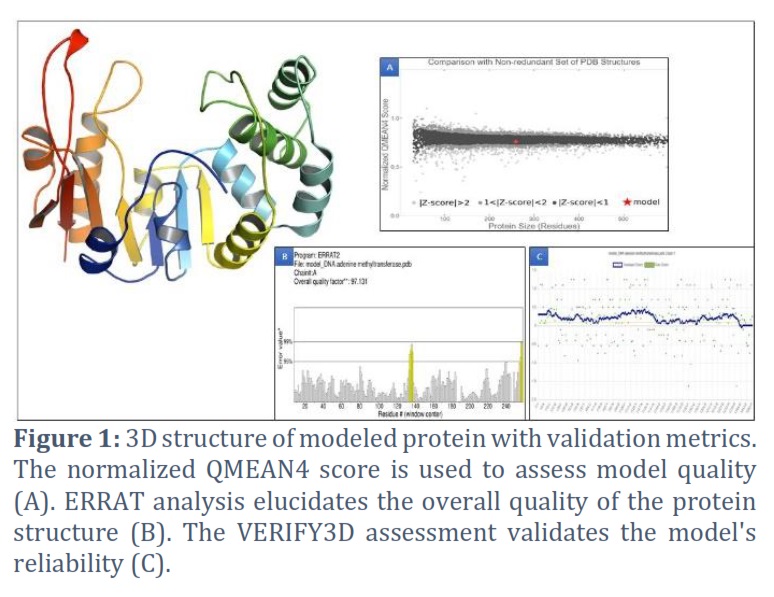
To validate the modeled structure, we initially conducted energy minimization. Subsequently, we uploaded the modeled structure to the SAVES server (http://nihserver.mbi.ucla.edu/SAVES/), which assessed its overall correctness using ERRAT and evaluated the stereochemical quality with PROCHECK. Additionally, we employed Verify3D to determine the compatibility of the atomic model with its corresponding amino acid sequence. The secondary structure and Ramachandran plot were generated using the PDBsum server. These comprehensive evaluations were performed to ensure the accuracy and reliability of the modeled structure.
Active site prediction
The active site of a protein is a distinct binding site that plays a crucial role in its specific function. Identifying these active sites is a fundamental step in structure-based drug design, as it helps determine potential targets for therapeutic interventions [14]. Precise prediction of active sites is an invaluable tool in the field of bioinformatics. In this study, we employed Discovery Studio software to predict the active site of the modeled protein. By utilizing this software, we were able to identify the key binding site, providing valuable insights for further docking analysis.
Natural compound library retrieval and preparation
‘Biopurify Phytochemicals’ subsets from the ZINC database, consisting of 2706 compounds, were downloaded in SDF format. These compounds were subjected to energy minimization using the ‘UFF’ force field and prepared by converting into pdbqt format for further docking-based virtual screening (VS).
Virtual screening
VS was performed with the PyRx 0.8 [15] software tools to evaluate the interaction of the natural compound library with the Dam protein. The binding affinity score was used as a metric for the evaluation.
Physicochemical properties and toxicity endpoints prediction
The top three selected compounds were subjected to evaluation of their physicochemical properties and predictions of various toxicity endpoints using the ProTox-II platform [16].
Results![]()
Initially, protein sequences of K. pneumoniae containing the Dam domain were retrieved from UniProt. Since there was no existing 3D structure available for this protein in the protein database, a 3D model was constructed using the SWISS-MODEL server. The structural properties of the Dam-containing protein in E. coli (PDB ID: 4RTL) were utilized as a template for the modeling process.
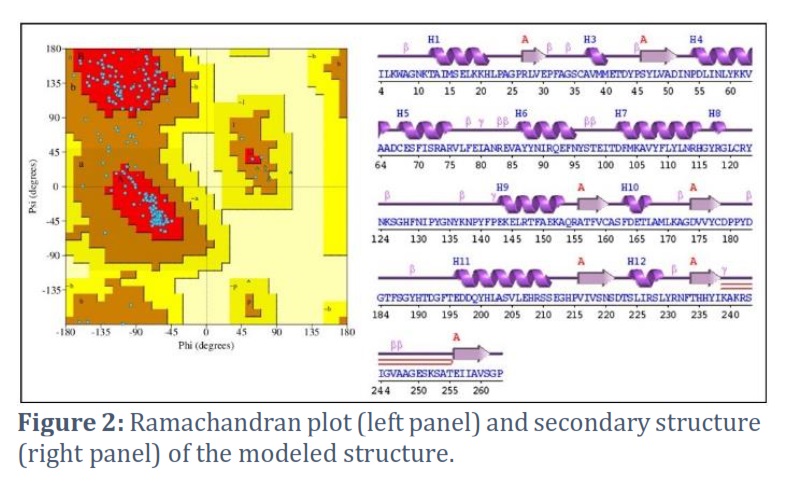
Several tools and web servers were used to validate the accuracy of the modeled structure. For this, the SAVES server and PDBsum were employed. According to PROCHECK analysis, 89.2% of the phi/psi angles of residues in the modeled structure were within the most favored regions, indicating a favorable stereochemical quality. Furthermore, the ERRAT graph produced a quality factor of 97.131, indicating a good model quality (a score greater than 50 is considered acceptable). The VERIFY3D server confirmed the model's high quality, with 95.24% of residues in the modeled protein scoring higher than 0.2. A normalized QMEAN4 score was also predicted, and VERIFY3D analysis revealed that 85.38% of the residues had an average 3D-1D score of >=0.1, indicating a "Pass" result for the structure (Figure 1).
PDBsum tool predicted the secondary structure of the modeled structure, which consisted of the secondary structure motif having;
- 1 sheet,
- 3 beta alpha beta units,
- 1 beta-hairpin,
- 7 strands,
- 12 helices,
- 12 helix-helix interactions,
- 17 beta turns, and
- 4 gamma turns
Additionally, a Ramachandran plot was also generated (Figure 2).
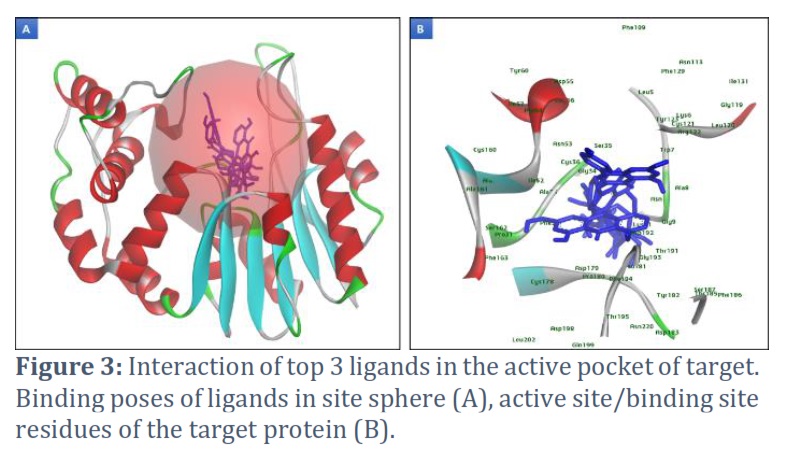
After validating the modeled protein structure and predicting its binding sites using DS 2021 software, we utilized the 'define and edit binding site' option. Through this process, it was determined that the best site had the attributes X=0.451893, Y=3.081089, and Z=90.425250. Subsequently, this site was utilized for the VS of prepared ligands, specifically natural compounds. We selected the top 10 hits for this study based on their binding affinity, which is listed in Table 1. Furthermore, we focused on the top 3 hits, providing a detailed description of their binding poses (Figure 3) within the active pocket and their interaction with active site residues.
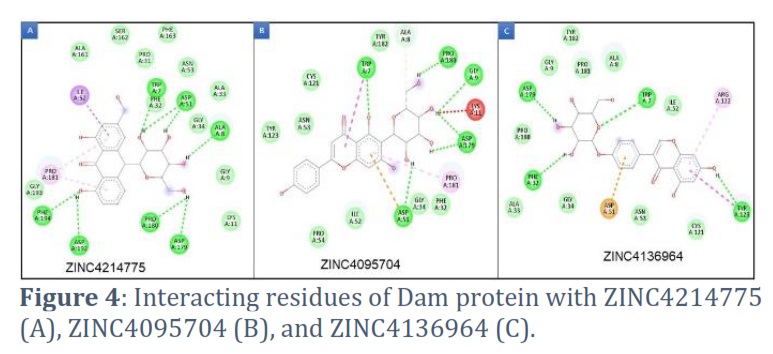
The detailed interaction analysis of the top 3 hit compounds is as follows. ZINC4214775 was found to interact with Asp192, Phe194, Gly193, Pro181, Ile52, Ala161, Ser162, Pro31, Phe163, Trp7, Phe32, Asn53, Asp51, Ala33, Gly34, Ala8, Gly9, Lys11, Asp179, and Pro180 residues of Dam. The Phe194, Asp192, Trp7, Asp51, Ala8, Asp179, and Pro180 residues formed H-bond with ZINC4214775 (Figure 4A). ZINC4095704 interacted with Tyr123, Asn53, Cys121, Trp7, Tyr182, Ala8, Pro180, Gly9, Lys11, Asp179, Pro181, Phe32, Gly34, Asp51, Ile52, and Pro54 residues of Dam. The Trp7, Pro180, Gly9, Asp179, and Asp51 residues H-bonded with ZINC4095704 (Figure 4B). Further, ZINC4136964 was found to interact with Asp51, Gly34, Phe32, Ala33, Pro180, Asp179, Gly9, Tyr182, Pro181, Ala8, Trp7, Ile52, Arg122, Tyr123, Cys121, and Asn53 residues of Dam. The Phe32, Asp179, Trp7, and Tyr123 residues formed H-bond with ZINC4136964 (Figure 4C).
The predicted values for these three hits indicate that they fall within an acceptable range for toxicity assessment, as shown in Figure 5. Additionally, the toxicity radar chart, which demonstrates the confidence of positive toxicity results compared to the average of its class, was generated. It reveals that these hits fall within an acceptable range, as depicted in Figure 6.
Figures & Tables
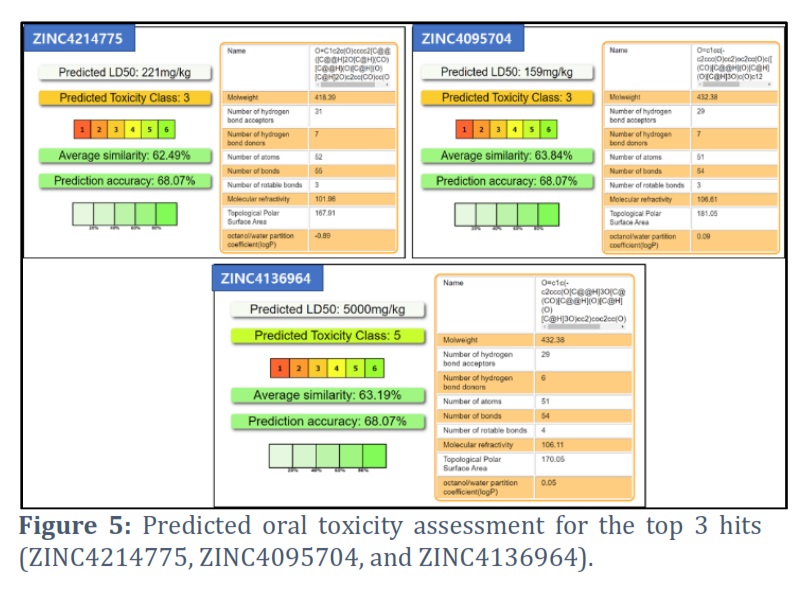
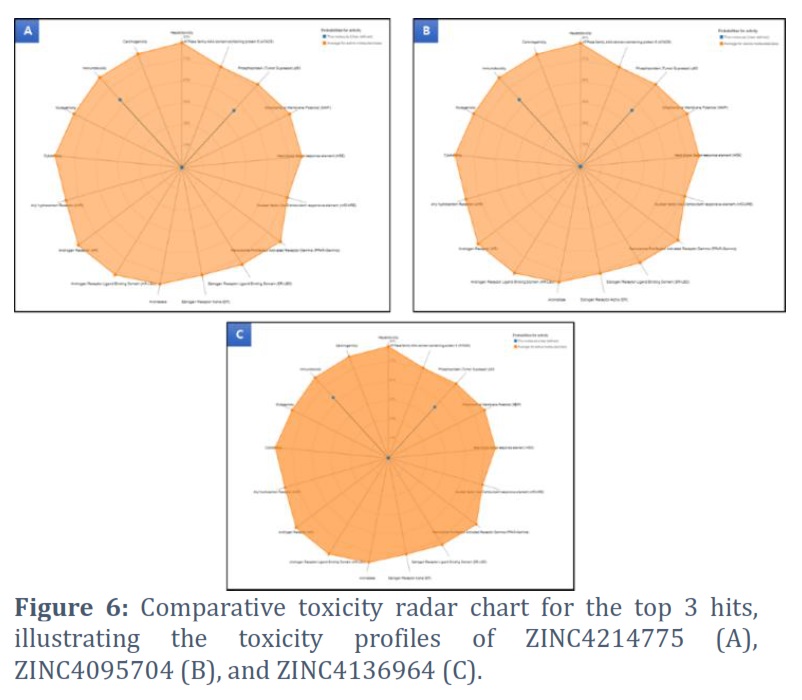
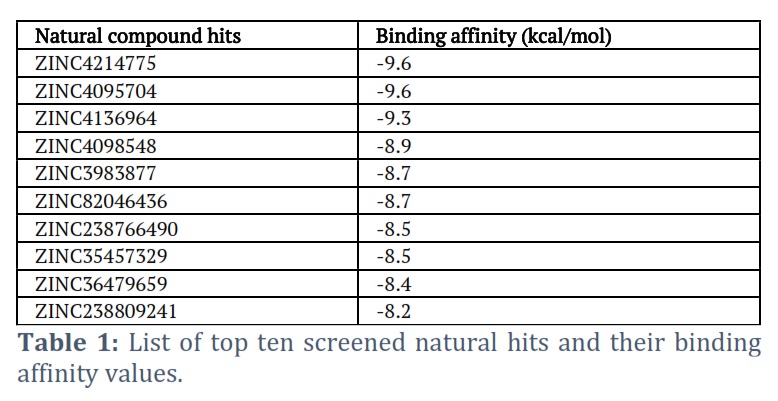
K. pneumoniae is a leading cause of hospital-acquired infections and is becoming increasingly drug-resistant [17]. The Dam is a protein that regulates genes associated with antibiotic resistance [8]. This study identified natural Dam inhibitors that could be used to combat antimicrobial resistance in K. pneumoniae. Among the identified compounds, this study focused on the top three hits (ZINC4214775, ZINC4095704, and ZINC4136964) that had strong binding with the DAM protein and interacted with active site residues of the DAM protein. Furthermore, we utilized the ProTox-II web tool to conduct toxicity assessments on the top 3 compounds. Prediction of compound toxicities plays a vital role in the drug design and development process, as it helps determine the safety profile of potential drugs. The ProTox-II server offers predictions for a wide range of toxicity endpoints, including acute toxicity, hepatotoxicity, cytotoxicity, carcinogenicity, mutagenicity, immunotoxicity, adverse outcome pathways, and toxicity targets. By utilizing these diverse techniques, ProTox-II provides comprehensive insights into the potentially toxic effects of compounds, facilitating the identification of safer and more effective drug candidates [16]. The three hits (ZINC4214775, ZINC4095704, and ZINC4136964) were found to have acceptable physicochemical properties and toxicity profiles. Using natural compounds in medicine discovery and the food industry has various advantages. These contain unmatched chemical diversity characterized by complicated structures and powerful biological activities. Natural products occupy a different region within the chemical space, complementing synthetic chemicals. Furthermore, the creation of libraries including natural product analogs has the potential to improve drug-like qualities. By enhancing the regulation of natural product biosynthesis, it becomes possible to investigate and comprehend targets and pathways involved in disease processes. Furthermore, using natural compounds allows for a more efficient transition from initial hits to medication development [18]. The hit compounds identified in this study are naturally occurring compounds.
This study focused on the Dam protein found in K. pneumoniae that encompasses a DNA methylation protein domain. Screening of phytochemicals against the Dam protein was performed to identify potential compounds of interest. Notably, ZINC4214775, ZINC4095704, and ZINC4136964 had higher affinity for the Dam protein and interacted with its active site residues. The predicted physicochemical properties and toxicity profiles of these compounds were found to be within an acceptable range. These compounds have the potential to act as Dam inhibitors and could be studied further for antimicrobial resistance management in K. pneumoniae.
Conflict of Interest
The authors declare no conflict of interest.
Conceptualization, Farah Anjum and Alaa Shafie; Methodology, Norah Ali Alshehri and Samia Musaad Almutairi; Writing original draft, Ameerah Ali Alshehri and Ayshah Musad Almutairi; Review & editing, Farah Anjum.
![]() References
References
- Martin RM, Bachman MA. Colonization, Infection, and the Accessory Genome of Klebsiella pneumoniae. Frontiers in Cellular and Infection Microbiology, (2018); 8: 4.
- Iredell J, Brown J, Tagg K. Antibiotic resistance in Enterobacteriaceae: mechanisms and clinical implications. The BMJ, (2016); 352h6420.
- Tesfa T, Mitiku H, Edae M, Assefa N. Prevalence and incidence of carbapenem-resistant K. pneumoniae colonization: systematic review and meta-analysis. Systematic Reviews, (2022); 11(1): 240.
- Heithoff DM, Sinsheimer RL, Low DA, Mahan MJ. An essential role for DNA adenine methylation in bacterial virulence. Science, (1999); 284(5416): 967-970.
- Robbins-Manke JL, Zdraveski ZZ, Marinus M, Essigmann JM. Analysis of global gene expression and double-strand-break formation in DNA adenine methyltransferase- and mismatch repair-deficient Escherichia coli. Journal of Bacteriology, (2005); 187(20): 7027-7037.
- Raghunathan N, Goswami S, Leela JK, Pandiyan A, Gowrishankar J. A new role for Escherichia coli Dam DNA methylase in prevention of aberrant chromosomal replication. Nucleic Acids Research, (2019); 47(11): 5698-5711.
- Westphal LL, Sauvey P, Champion MM, Ehrenreich IM, Finkel SE. Genomewide Dam Methylation in Escherichia coli during Long-Term Stationary Phase. mSystems, (2016); 1(6): e00130-16.
- Adam M, Murali B, Glenn NO, Potter SS. Epigenetic inheritance based evolution of antibiotic resistance in bacteria. BMC Ecology and Evolution, (2008); 8: 52.
- Pan SY, Zhou SF, Gao SH, Yu ZL, Zhang SF, et al. New Perspectives on How to Discover Drugs from Herbal Medicines: CAM's Outstanding Contribution to Modern Therapeutics. Evidence-Based Complementary and Alternative Medicine, (2013); 2013627375.
- Baig MH, Ahmad K, Rabbani G, Danishuddin M, Choi I. Computer Aided Drug Design and its Application to the Development of Potential Drugs for Neurodegenerative Disorders. Current Neuropharmacology, (2018); 16(6): 740-748.
- Bassani D, Moro S. Correction: Bassani, D.; Moro, S. Past, Present, and Future Perspectives on Computer-Aided Drug Design Methodologies. Molecules 2023, 28, 3906. Molecules, (2023); 28(13): 3906.
- Kumar S. Molecular modeling and identification of substrate binding site of orphan human cytochrome P450 4F22. Bioinformation, (2011); 7(4): 207-210.
- Horton JR, Zhang X, Blumenthal RM, Cheng X. Structures of Escherichia coli DNA adenine methyltransferase (Dam) in complex with a non-GATC sequence: potential implications for methylation-independent transcriptional repression. Nucleic Acids Research, (2015); 43(8): 4296-4308.
- Fauman EB, Rai BK, Huang ES. Structure-based druggability assessment–identifying suitable targets for small molecule therapeutics. Current Opinion in Chemical Biology, (2011); 15(4): 463-468.
- Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods in Molecular Biology, (2015); 1263: 243-250.
- Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Research, (2018); 46(W1): W257-W263.
- Al Bshabshe A, Al-Hakami A, Alshehri B, Al-Shahrani KA, Alshehri AA, et al. Rising Klebsiella pneumoniae Infections and Its Expanding Drug Resistance in the Intensive Care Unit of a Tertiary Healthcare Hospital, Saudi Arabia. Cureus, (2020); 12(8): e10060.
- Lam KS. New aspects of natural products in drug discovery. Trends in Microbiology, (2007); 15(6): 279-289.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0