

Full Length Research Article
Plausible inhibitors of malaria parasite Plasmodium falciparum 3D7 ATP-dependent DNA helicase
Mohammad Othman Alkurbi
Adv. life sci., vol. 10, no. 4, pp. 644-650, December 2023
*– Corresponding Author: Mohammad Othman Alkurbi (mokurbi@uqu.edu.sa)
Authors' Affiliations
[Date Received: 14/07/2023; Date Revised: 05/08/2023; Date Published Online: 31/12/2023; Date Updated:09/09/2025]
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Plasmodium falciparum is a parasite (protozoan) of humans, and the lethal species of Plasmodium that causes malaria in humans. In the lack of a medically validated malaria vaccine, there are merely a few inexpensive medications available for therapy. Studies of diverse enzymes used in pharmaceutical drug discovery have become essential aspects. The theoretical analysis helps to screen novel drug candidates.
Methods: Here, we have optimized three biologically active compounds, netropsin, nogalamycin, and novobiocin, and also carried out a molecular docking study with the protein ATP-dependent DNA helicase (UvrD) from Plasmodium falciparum 3D7.
Results: The plasmoDB id of the designated protein is PF3D7-0514100. Our calculations show that netropsin, nogalamycin, and novobiocin can have an affinity for Plasmodium falciparum.
Conclusion: Our study also predicted that novobiocin would give a better result with this protein than netropsin and nogalamycin. The frontier molecular orbitals and electrostatic potential (MEP) maps also support the higher activity of the novobiocin compound.
Keywords: Plasmodium falciparum; Netrropsin; Nogalamycin; Novobiocin; HOMO-LUMO; MEP & Prankweb-2 platform
Introduction![]()
Malaria is a disease that has gained international attention and poses serious health and socioeconomic issues in more than 100 different nations. Four species of Plasmodium (P. falciparum, P. ovale, P. malariae, and P. vivax) cause malaria in humans [1]. Plasmodium falciparum is the most virulent and lethal species that frequently infects people. It is responsible for 95 percent of malaria deaths. Several parasite proteins could be effective antimalarial therapeutic targets [2,3]. Helicase inhibitors may be a feasible approach for developing novel therapeutics because helicases are essential for the metabolism of DNA and RNA [4]. Enzyme mobility may be hampered by the development of a small molecule (compound) DNA complex, which may cause the suppression of enzyme activities. The anti-helicase activity of various DNA intercalating agents was evaluated [5]. The researchers utilized three types of nucleic acid-interacting compounds in their research: (1) Agents that intercalate DNA, such as nogalamycin and daunorubicin, (2) minor groove binders like distamycin and netropsin, and (3) non-intercalating topoisomerase inhibitors like novobiocin and camptothecin.
Streptomyces produces a tiny molecule, which is netropsin. Small molecules that bind to the minor groove of DNA have considerable significance for developing medicinal applications such as gene expression regulation and the creation of possible anticancer, antibiotic, and antiviral medicines [6-8]. Netropsin has been demonstrated to be effective against both Gram-positive and Gram-negative bacteria [9]. Netropsin, a naturally occurring peptide derived from S. netropsis in 1951, was one of the first small molecules found to bind specifically to the minor groove of particular DNA sequences [10]. Netropsin serves as the foundation for various alternatives that have been synthesized [11], including combinatorially, to study and alter ligand-DNA interactions [8,12-16]. Based on X-ray crystallographic structures, netropsin interacts deep in the minor grooves of DNA primarily with AT-rich stretches of double-stranded DNA via Van der Waals, hydrogen bonding, and electrostatic interactions [12,17-18]. Compared to other AT-rich sequences studied, netropsin interacts with DNA particularly well at duplexed AATT locations [13-16,19].
The Type II topoisomerases [20] are a class of widely distributed ATP-dependent enzymes that includes DNA gyrase [20-21]. The majority of DNA-related processes in biology have been linked to these enzymes. The coumarins and cyclothialidines are competitive inhibitors, according to steady-state kinetic investigations of the hydrolysis reaction of the integral A292 DNA gyrase molecule [22-25]. Novobiocin, clorobiocin, and the coumermycin are among the members of the coumarin family of naturally occurring antibiotics. Novobiocin is made up of a benzoic acid moiety that has been isopentenylated and is connected to hydroxycoumarin by an amide, which is then attached to the sugar noviose [26]. The Streptomyces strain S. sphaeroides encodes two gyrase B proteins: one that is novobiocin-sensitive and a second, novobiocin-resistant protein that is only expressed during times of metabolic stress when novobiocin production is also induced. Given how little information there is about coumarin-resistant mutations, exploring the sequences (amino acids) of these two proteins tells an unexpectedly large number of variations [27].
Bhuyan and Dietz discovered nogalamycin for the Upjohn corporation in 1965. Nogalamycin is a drug made by the bacterium Streptomyces nogalater [28]. Although significant toxicity impeded its advancement to clinical trials, it was found to show excellent effectiveness against Gram-positive bacteria and numerous cancer cell lines. In the late 1970s, menogaril, a semi-synthetic derivative, was created [29]. It has considerably more potent anticancer properties and lacks nogalamycin’s considerable acute toxicity. Despite these encouraging outcomes, phase II of its clinical trials saw the end of its development [30]. Nogalamycin, an anti-cancer anthracycline, intercalates itself into the DNA double helix. L-nogalose and L-nogalamine, two carbohydrate units that interact with the DNA grooves (minor and major), respectively, promote the binding. Anthracyclines are naturally occurring microbiological compounds with medical value that are frequently used to treat cancer [31]. The growing interest in such types of diseases motivated us to explore the interactions between the target protein (UvrD) Plasmodium falciparum 3D7 (PlasmoDB: PF3D7-0514100) with netropsin, nogalamycin, and novobiocin.
Methods![]()
All three compounds, i.e., netropsin (1), nogalamycin (2), and novobiocin (3), were optimized at 298.15 K in Gaussian16 software [32]. Calculations were done on all the compounds using functional M06-2X [33] with basis sets 6-311G [34-35] DFT/M06/6-311G level of theory in the investigations of the interactions between the target protein UvrD (ATP-dependent DNA helicase) , Plasmodium falciparum 3D7, with PlasmoDB no. PF3D7-0514100 and compounds 1-3 were performed using the AutoDock Vina program and AutoDock-Tools (ADT) [36-37]. The protein was modeled by using the I-Tasser platform. The protein’s binding site was predicted using the Prankweb-2 platform [38]. All the protein’s water molecules were removed before the docking calculation. When allocating both grid vatedbox sizes (30x30x30Å concentrated at X = 148.076, Y = 141.265, Z = 97.964), the residues of each protein’s active site were considered. The biologically active compounds, i.e., 1, 2 and 3, which are geometrically optimized, and the compound-protein interactions and also docked poses were imagined using the Biovia Discovery Studio Visualizer 2021 [39].
Results![]()
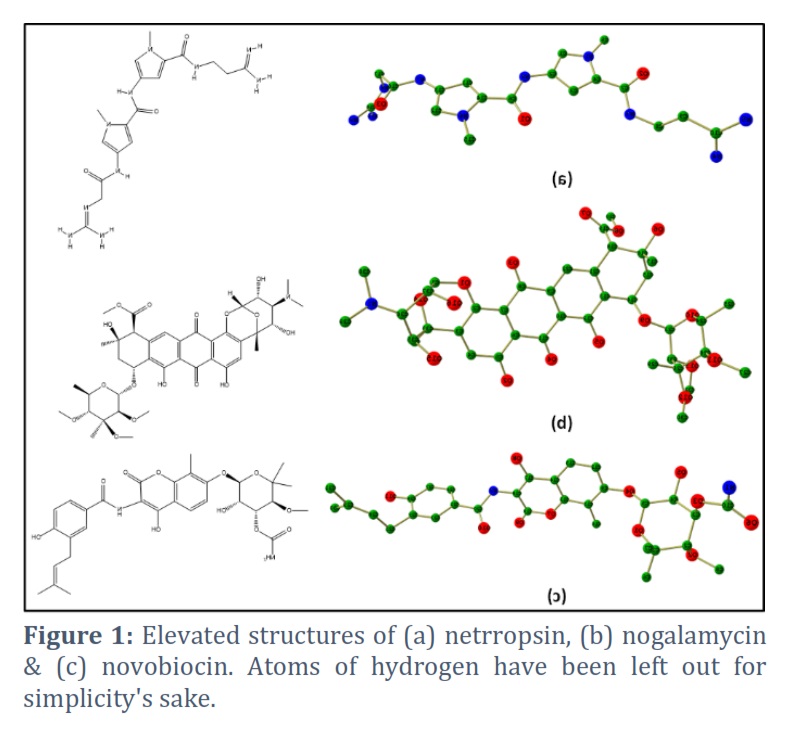
Optimized structure of compounds 1-3
We have conducted a thorough computational study for compounds 1-3 to get more insights. Detailed optimization calculations have been attempted on the compounds to gain in-depth knowledge and understand their properties for the aimed applications. Initially, we took compound one and performed optimization, followed by compounds 2-3. The optimized structure of compounds 1-3 is shown in Figure 1.
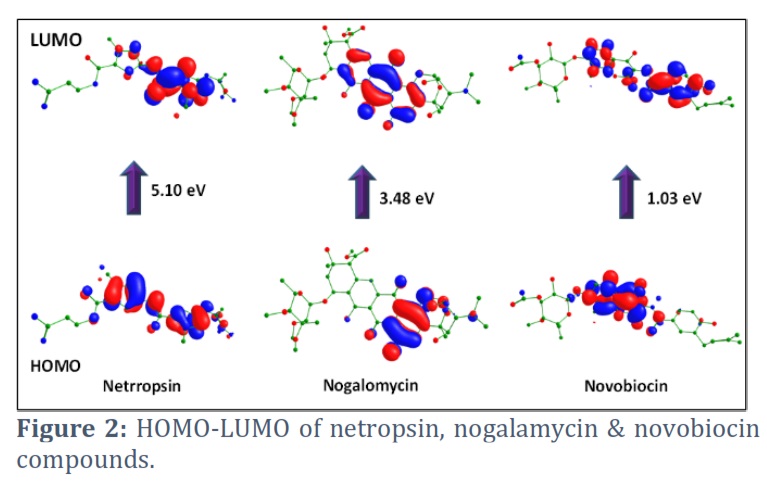
Frontier molecular orbitals (HOMO-LUMO)
Exploration of FMO using computational methods has become very popular and is a widely used tool to investigate the molecular reactivity of organic compounds. The properties of frontier orbitals are fundamental in quantum chemistry. They are instrumental in exploring different properties of molecules, like electronic properties, optical properties, and the charge transfer taking place within the molecule. The electronic absorption occurring within the molecule is due to the transfer of an electron from HOMO to LUMO. The HOMO orbital can give an electron and describes the ionization potential, while the LUMO performs as an electron acceptor and describes the electron affinity. The frontier molecular orbitals (FMO) analysis of all the molecules was carried out at the same level as that of the optimization. HOMO-LUMO of 1-3 is revealed in Figure 2. The energy gap between HOMO-LUMO is a very crucial parameter that can be exploited to investigate the chemical reactivity of the molecule. Further, this difference represents the electronic excitation energy. Because these orbitals' energies are the closest to one another among all orbitals of different energy levels, the HOMO-LUMO gap is where excitations are most likely to occur. As the HOMO-LUMO Gap converges, excitations become easier in the molecule. Also, a smaller HOMO-LUMO gap corresponds to better stability, while a larger HOMO-LUMO gap is always associated with higher kinetic stability and lower chemical reactivity. The gap can also be exploited to have an idea about the biological activity of the concerned molecule. Here, we have computed frontier orbitals of Netropsin, Nogalamycin, and Novobiocin and found the lesser HOMO-LUMO energy gap in Novobiocin, which can show more binding with the target protein ATP-dependent DNA helicase (UvrD), Plasmodium falciparum 3D7.
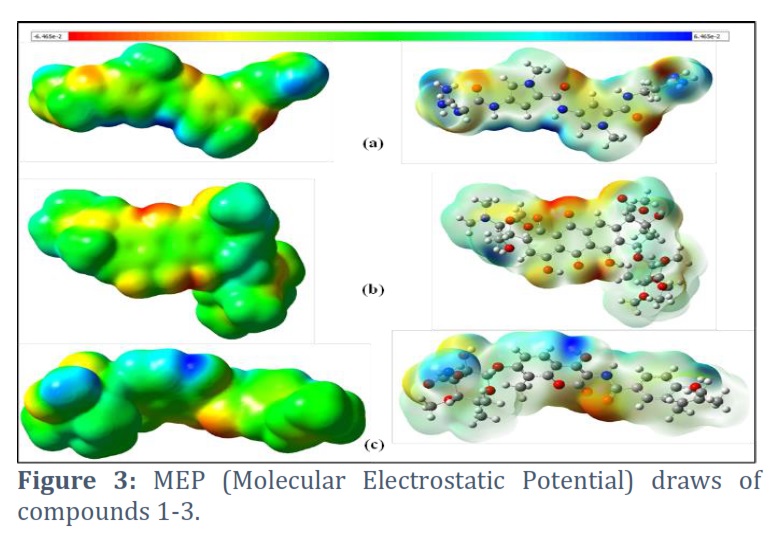
Molecular electrostatic potential (MEP)
The MEP (molecular electrostatic potential) plot is a pictorial technique to figure out the charge distribution region for nucleophilic and electrophilic attack. The red color region indicates the maximum negative potential, whereas the most significant positive potential is at the blue region. The green and yellow areas of the molecular electrostatic potential surfaces are between the two extremes. The red region is prone to electrophilic attack, whereas the blue region is for nucleophilic attack. The molecular electrostatic potential plots of compounds (1-3) are shown in Figure 3. Several reactive centers are present in all three compounds. The red region is found nearer to the oxygen atoms, whereas the blue is at the nitrogen atoms. Figure 3 also shows that relatively more nucleophilic sites are present in compound 3.
Molecular Docking Analysis
Protein–ligand docking is a potent and popular computational technique that is used to predict the conformation and orientation (pose) of a ligand bound to a protein. Docking has made the visualization of potential interactions of ligands with proteins easy and efficient. Molecular docking has become a famous computational tool to examine, substantiate, and probe the different possible conformations of proteins. It is widely used to study protein-ligand interaction, drug development, prediction of bound conformation, and calculation of the binding free energy of ligand to the target protein. Basically, the docking software is the interplay between the search algorithm and the scoring function. The role of the former is to find different conformations for the ligand, while the latter is used to rank the different conformations obtained by the search algorithm.
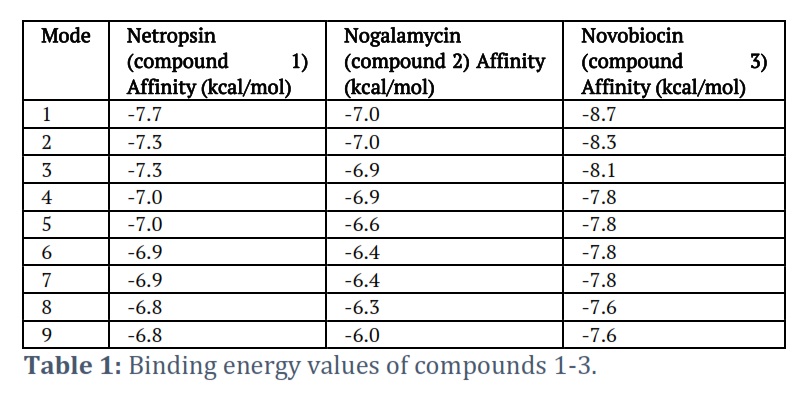
Here, to examine the binding affinities and types of intermolecular bonding interactions between the respective compounds and the target proteins, the docked models were analyzed. The various docking postures and energies were observed and assessed. The binding energy values obtained from the docking result of Netropsin are -7.7, -7.3, -7.3, -7.0, -7.0, -6.9, -6.9, -6.8, and -6.8 kcal/mol, respectively. The docking pose with the highest negative binding energy has the highest binding affinity. Significant interactions like H-bonding and Pi-alkyl were present. The H-bonding is present at a distance of 1.80 Å. Two dominant Pi-alkyl interactions are also present at a distance of 4.63 Å and 4.86 Å (see Figure 4). The binding energy values obtained from the docking results of nogalamycin and novobiocin with the highest binding affinity are -7.0 and -8.7 kcal/mol, respectively (see Figures 5A and 5B).
The binding energy values for compounds 1-3 are summarized in Table 1. The crucial interactions between compound two and the amino acid residues are H-bonding, pi-alkyl, and pi-sigma, which are at a distance of 2.78 Å, 5.33 Å, and 3.60 Å, respectively. Consistently, the dominant interactions present in compound three are H-bonding, pi-pi, pi-alkyl, and pi-sigma, which are present at a distance of 2.98 Å, 5.22 Å, 5.09 Å, and 3.61 Å, respectively.
Figures & Tables
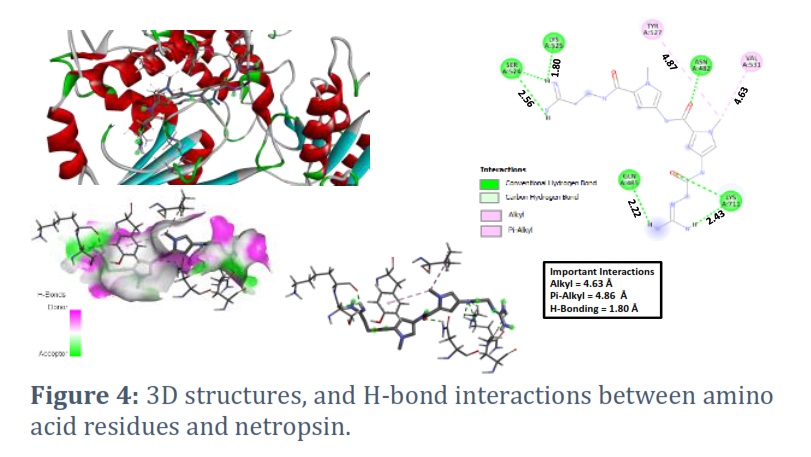
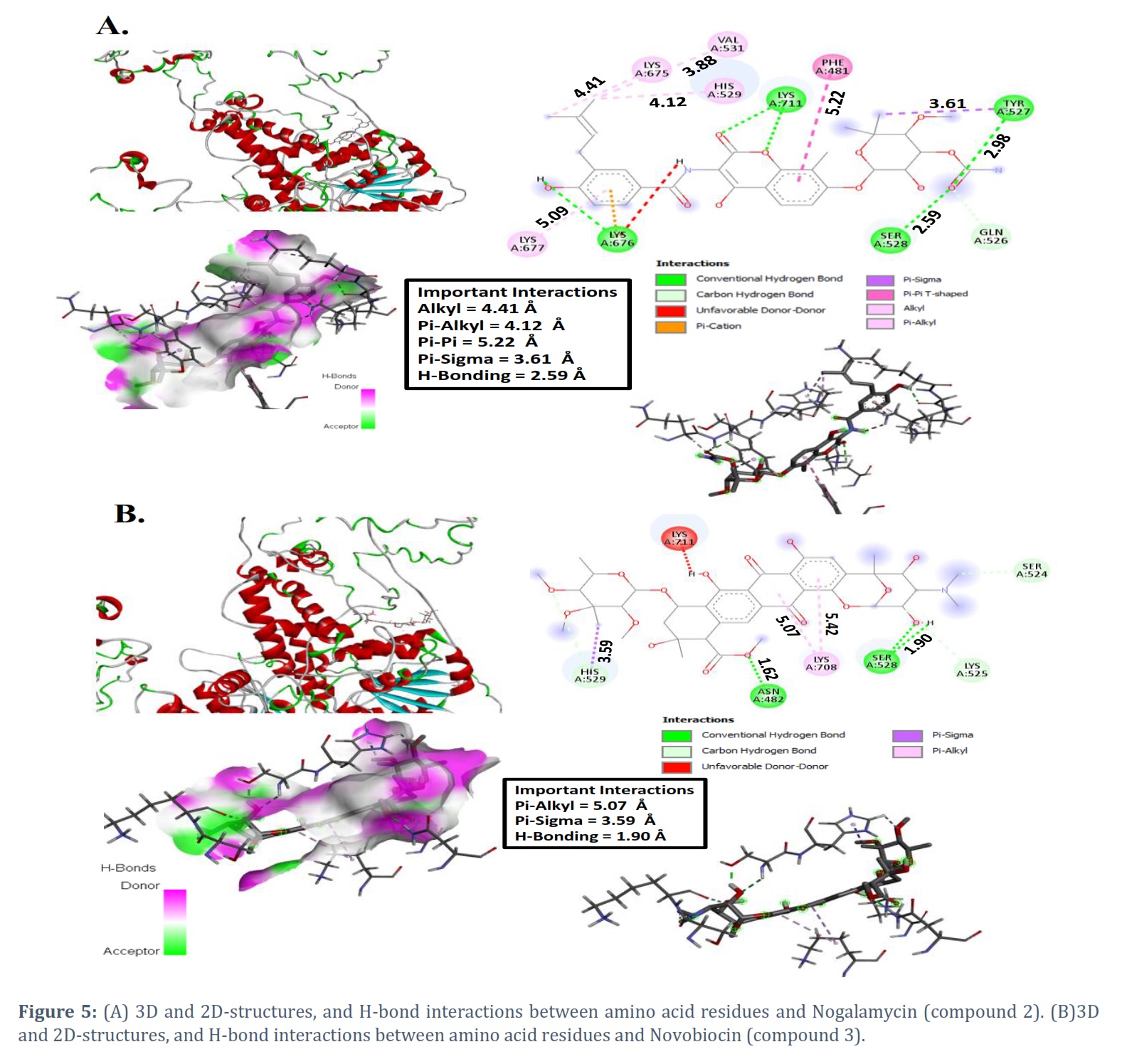
In this study, our main focus is to study the plausible inhibitors of malaria parasite Plasmodium falciparum 3D7 ATP-dependent DNA helicase. From the previous reports, netropsin, nogalamycin, and novobiocin compounds play a vital role in biological studies [8,9,18,26,31]. To confirm the stability of these compounds, first, we have optimized Netropsin, nogalamycin, and novobiocin compounds to get a stable geometry by using the DFT method. Furthermore, we have also performed the molecular docking of these compounds against the protein malaria parasite Plasmodium falciparum 3D7 ATP-dependent DNA helicase to figure out their activity. Activity can be found by calculating their binding scores (kcal/mol). The binding energy of netropsin compound against the protein is found as -7.7, -7.3, -7.3, -7.0, -7.0, -6.9, -6.9, -6.8, and -6.8 kcal/mol, respectively, and these are selected for further analysis based on energy score and hydrogen bond interactions. The netropsin compound formed hydrogen bonds with seven residues: ASN482, GLN485, SER524, LYS525, TYR527, VAL531, and LYS711 (see Figure 4). Further, molecular docking showed that the binding energy of the nogalamycin compound against the protein was -7.0, -7.0, -6.9, -6.9, -6.6, -6.4, -6.4, -6.3, and -6.0 kcal/mol, respectively. The nogalamycin compound formed hydrogen bonds with 10 residues PHE481, GLN526, TYR527, SER528, HIS529, VAL531, LYS675, LYS676, LYS677 and LYS711 (see Figure 5).
Molecular docking study observed the binding energy of the nogalamycin compound against the protein as -8.7, -8.3, -8.1, -7.8, -7.8, -7.8, -7.8, -7.6, and -7.6 kcal/mol, respectively. This compound formed hydrogen bonds with 7 residues ASN482, SER524, LYS525, SER528, HIS529, LYS708 and LYS711 (see Figure 5).
Among the three compounds (1-3), one with the most negative binding energy value will have the highest critical potential. However, the docked complexes' minimal comparative interaction energies were established to be -7.7, -7.0, and 8.7 kcal/mol for compounds 1-3, respectively. These negative values indicate higher protein binding potential. Compound 2 has a lower interaction energy, proving lesser efficiency in binding to the protein, while compound 3 has a higher binding energy, demonstrating a higher efficiency in binding with the protein. It has been found that hydrogen bonding has played a crucial role in connecting compounds by proteins. These three molecules revealed an excellent binding affinity and stable binding modes as predicted by molecular docking. Furthermore, the frontier molecular orbitals (HOMO-LUMO) indicated that electrons may transfer relatively faster in novobiocin than in netropsin and nogalamycin. Molecular electrostatic potential maps also predicted that novobiocin may have higher reactivity. Ultimately, our findings directly relate to the community working in biological studies and the related interface of medicinal chemistry.
The author declares that there is no conflict of interest.
![]() References
References
- Snow RW, Guerra CA, ANoor AM, Myint HY, Hay SI. The Global Distribution of Clinical Episodes of Plasmodium falciparum Malaria. Nature, (2005); 434 (7030): 214-7.
- Carlton JM, Angiuoli SV, Suh BB, Kooij TW, Pertea M, et al. Genome Sequence and Comparative Analysis of the Model Rodent Malaria Parasite Plasmodium Yoelii Yoelii. Nature, (2002); 419 (6906): 512-9.
- Gardner MJ, Hall N, Fung E, White O, Berriman M, et al. Genome Sequence of the Human Malaria Parasite Plasmodium falciparum. Nature, (2002); 419 (6906); 498-511.
- Tuteja R. Helicases: Feasible Anti-malarial Drug Target for Plasmodium falciparum. FEBS J, (2007); 274 (18): 4699-704.
- Mehta J, and Tuteja R. A Novel Dual Dbp5/DDX19 Homologue from Plasmodium falciparum Requires Q motif for Activity. Mol. Biochem. Parasitol, (2011); 176 (1): 58-63.
- White S, Szewczyk JW, Turner JM, Baird EE, Dervan PB. Recognition of the four Watson-Crick base pairs in the DNA minor groove by synthetic ligands. Nature, (1998); 391 (6666): 468.
- Chenoweth DM, Harki DA, Phillips JW, Dose C, Dervan PB. Cyclic pyrrole-imidazole polyamides targeted to the androgen response element. J. Am. Chem. Soc., (2009); 131 (20): 7182-7188.
- Schneider S, Keller S, Wolter FE, Röglin L, Beil W, et al. Proximicins A, B, and C-antitumor furan analogues of netropsin from the marine actinomycete Verrucosispora induce upregulation of p53 and the cyclin kinase inhibitor p21. Angew. Chem. Int. Ed., (2008), 47 (17): 3258-3261.
- Zimmer C, Wähnert U. Nonintercalating DNA-binding ligands: specificity of the interaction and their use as tools in biophysical, biochemical and biological investigations of the genetic material. Prog. Biophys. Molec. Biol., (1986); 47 (1): 31-112.
- Finlay AC, Hochstein FA, Sobin BA, Murphy FX. Netropsin, a New Antibiotic Produced by a Streptomyces. J. Am. Chem. Soc., (1951); 73 (24): 341-343.
- Boger DL, Fink BE, Hedrick MP. Total Synthesis of Distamycin A and 2640 Analogues: A Solution-Phase Combinatorial Approach to the Discovery of New, Bioactive DNA Binding Agents and Development of a Rapid, High-Throughput Screen for Determining Relative DNA Binding Affinity or DNA Binding Sequence Selectivity. J. Am. Chem. Soc., (2000); 122 (27): 6382-6394.
- Neidle S. DNA minor-groove recognition by small molecules. Natural Product Reports, (2001); 18(3): 291-309.
- Lewis EA, Munde M, Wang S, Rettig M, Le, V Machha V, Wilson WD. Complexity in the binding of minor groove agents: netropsin has two thermodynamically different DNA binding modes at a single site. Nucleic Acids Res., (2011); 39 (22): 9649-9658.
- Freyer MW, Buscaglia R, Nguyen B, Wilson WD, Lewis EA. Binding of netropsin and 4,6-diamidino-2-phenylindole to an A2T2 DNA hairpin: A comparison of biophysical techniques. Anal. Biochem., (2006); 355(2): 259-266.
- Lah J, Drobnak I, Dolinar M, Vesnaver G. What drives the binding of minor groove-directed ligands to DNA hairpins? Nucleic Acids Res., (2008); 36 (3): 897-904.
- Ramos JP, Le VH, Lewis EA. Role of Water in Netropsin Binding to an A2T2 Hairpin DNA Site: Osmotic Stress Experiments. J. Phys. Chem. B, (2013): 117 (50): 15958-15965.
- Goodwin KD, Long EC, Georgiadis MM. A host–guest approach for determining drug–DNA interactions: an example using netropsin Nucleic Acids Res., (2005); 33 (13): 4106-4116.
- Hecke KV, Nam PC, Nguyen MT, Meervelt LV. Netropsin interactions in the minor groove of d(GGCCAATTGG) studied by a combination of resolution enhancement and ab initio calculations. FEBS J., (2005); 272 (14): 3531-3541.
- Freyer MW, Buscaglia R, Cashman D, Hyslop S,Wilson WD, Chaires JB, Lewis EA. DNA conformational effects on the interaction of netropsin with A-tract sequences. Biophys. Chem., (2007); 126 (1-3): 186-196.
- Wang JC. DNA topoisomerases. Ann. Rev. Biochem., (1985); 54, 665-697.
- Reece RJ, Maxwell A. DNA gyrase: structure and function. Crit.Rev. Biochem. Mol. Biol., (1991); 26 (3-4): 335-375.
- Sugino A, Cozzarelli NR. The intrinsic ATPase of DNA gyrase. J.Biol. Chem., (1980); 255 (13):6299-6306.
- Sugino A, Higgins NP, Brown PO, Peebles CL, Cozzarelli NR. Energy Coupling in Dna Gyrase and The Mechanism of Action of Novobiocin. Proc. Natl Acad. Sci., (1978): 75 (10): 4838-4842.
- Staudenbauer WL, Orr E. DNA Gyrase: Affinity Chromatography on Novobiocin-Sepharose and Catalytic Properties. Nucleic Acids Res., (1981); 9 (15): 3589- 3603.
- Nakada N, Gmuender H, Hirata T, Arisawa M. Mechanism of Inhibition of DNA Gyrase by Cyclothialidine, A Novel DNA Gyrase Inhibitor. Antimicrob. Agents Chemother., (1994); 38 (9): 1966-1 973.
- Kuo MS, Yurek DA, Chirby DG, Cialdella J, Marshall VP. Microbial 0-carbamoylation of novobiocin. J. Antibiof., (1991); 44 (10): 1096-1100.
- Thiara AS, Cundliffe E. Expression and analysis of 2 gyr8 genes from the novobiocin producer, Streptomyces sphaeroides. Moi. Microbioi., (1993); 8 (3): 495-506.
- Bhuyan BK, Dietz A. Fermentation, taxonomic, and biological studies on nogalamycin. Antimicrob. Agents Chemother., (1965); 5, 836 – 844.
- Wiley PF, Johnson JL, Houser DJ. Nogalamycin analogs having improved antitumor activity. J. Antibiot., (1977); 30 (7): 628-629.
- Nogalamycin. ChemicalBook.com. Accessed November 28, 2012.
- Metsä-Ketelä M, Niemi J, Mäntsälä P, Schneider in Topics in Current Chemistry, Anthracycline Chemistry and Biology I: Biological Occurence and Biosynthesis, Synthesis and Chemistry, (Ed.:Krohn K), Springer-Verlag, Berlin/Heidelberg (2008); 282 101–140.
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, et al. Gaussian 16, Revision C.01; Wallingford CT, USA, 2016.
- Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc., (2008); 120 (1-3): 215-241.
- McLean AD, Chandler GS. Contracted Gaussian-basis sets for molecular calculations. 1. 2nd row atoms, Z=11-18. J. Chem. Phys., (1980); 72, 5639-5648.
- Krishnan R, Binkley JS, Seeger R Pople JA. Self-Consistent Molecular Orbital Methods. 20. Basis set for correlated wave-functions. J. Chem. Phys., (1980); 72, 650-654.
- Trott O, Olson AJ, Vina A. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comp. Chem., (2010); 31 (2): 455-461.
- Roy A, Kucukural A, Zhang Y. I-TASSER: A Unified Platform for Automated Protein Structure and Punction Prediction. Nat Protoc., (2010); 5 (4):725-738.
- Jakubec D, Skoda P, Krivak R, Novotny M, Hoksza D, PrankWeb 3: Accelerated Ligand-Binding Site Predictions for Experimental and Modelled Protein Structures, Nucleic Acids Res., (2022); 50 (W1): W593-W597.
- BIOVIASystemes, Dassault. [BIOVIA Discovery Studio]. San Diego: Dassault Systemes. (2021).
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0