

Full Length Research Article
First report of Tomato leaf curl New Delhi virus (a bipartite begomovirus) from round gourd plant in Swat region of Pakistan: its phylogenetic and biogeographic analysis
Chand Bibi1, Raham Sher Khan1*, Murad Ali Rahat2, Asadullah3, Muhammad Zahoor4, Hassan Sher5, Khadim Hussain6, Fazal Akbar2*, Mohammed Ali Al-Saleh6
Adv. life sci., vol. 11, no. 2, pp. 398-405, May 2024
*– Corresponding Author: Fazal Akbar (fazalakbar@uswat.edu.pk); Raham Sher Khan (rahamsher@awkum.edu.pk)
Authors' Affiliations
2. Center for Biotechnology and Microbiology University of Swat, KP – Pakistan
3. Department of Biotechnology and Genetic Engineering, Hazara University, Mansehra, KP – Pakistan
4. Department of Biochemistry, University of Malakand, KP – Pakistan
5. Centre for Plant Science and Biodiversity, University of Swat – Pakistan
6. Chair of Date Palm Research, Plant Protection Department, College of Food and Agriculture Sciences, King Saud University – Saudi Arabia
[Date Received: 17/08/2023; Date Revised: 28/01/2024; Date Available Online: 18/04/2024]
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: The family Geminiviridae contains plant viruses commonly referred to as geminiviruses, which cause diseases in numerous important plants. Geminiviridae is one of the most important family of plant-infecting viruses that has been divided into fourteen recognized genera. Amongst these, the genus Begomovirus is the largest and economically most important one, viruses of the genus are further categorized into bipartite (two genomic components called DNA-A and DNA-B) and monopartite (single genomic component or DNA-A) begomoviruses.
Method: The genomic DNA was extracted from symptomatic plant samples and both the DNA-(B and A) components of a bipartite begomovirus were amplified via rolling circular amplification (RCA) and PCR. The amplified DNA-A and DNA-B were cloned and sequenced by Primer walking, Sanger sequencing, and Illumina sequencing methods. The phylogenetic and biogeographic analyses of both components were performed by various bioinformatics tools.
Results: DNA-A (accession number MW722701) and DNA-B (accession number MW722782) of begomoviruses that were sequenced shared >95% sequence similarity with the bipartite begomovirus known as Tomato Leaf Curl New Delhi virus (TLCuNDV). Phylogenetic and biogeographic analyses proposed several possible ancestors, and multiple dispersal and vicariance events may be involved in the evolution of this virus.
Conclusions: This is the first time a bipartite begomovirus has been found to infect a plant in the study area (District Swat, Pakistan). The existence of ToLCNDV may pose a major danger to other important crops in the region.
Keywords: Bipartite Begomoviruses; Phylogeny; Biogeography; BLAST; S-DIVA; BBM; DEC
Introduction![]()
Plant viruses, like other viruses, are obligatory intracellular parasites that depend only on the resources of their host cells to replicate. Serious plant illnesses are caused by plant viruses that infect plants. Plant viruses are encased in a capsid protein known as the protein coat and have their genetic elements either in RNA or DNA form. The bulk (>90%) of viruses that infect plants have RNA as their genetic material. Only around 10% of plant viruses have DNA as their genetic material. Geminiviridae, Nanoviridae, and Caulimoviridae are the only three families of DNA viruses that infect plants and are further divided into two types. The single stranded (ssDNA) viruses with circular genomes are placed in the families Nanoviridae and Geminiviridae, viruses of both of these families replicate by a rolling circular mechanism. The genomes of viruses of the Caulimoviridae family are made of double-stranded (ds) DNA, and they replicate by reverse transcription [1, 2].
The second largest family of viruses that infect plants is called Geminiviridae [3]. Members of this family are highly versatile as they infect both monocot and dicot host plants. These viruses are transmitted by insect vectors and have a short circular ssDNA genome of 2.5 to 3.0 kb [4]. Leaf twisting (either upward or downward), deformation, stunted growth, discoloration, reduction in leaf size, discoloration of leaf veins, enation (leaf-like outgrowth arising main leaf), decrease in fruit and vegetable size and also reduction in plant yield, abortion of flowers, fruit discoloration, and vegetable shape change are typical disease symptoms of geminiviruses in plants. Numerous plant hosts are afflicted by illnesses caused by geminiviruses, which lead to reduced yield and significant financial loss. Begomovirus, Becurtovirus, Citlodavirus, Capulavirus, Curtovirus, Eragrovirus, Grablovirus, Mulcrilevirus, Mastrevirus, Maldovirus, Opunvirus, Turncurtovirus, Topocuvirus, and Topilevirus are the fourteen recognized genera of the Geminiviridae family. This categorization is based on the genetic makeup of the virus, the kind of whitefly vector that spread the viruses, and the vast host range of the virus. The primary and most well researched genus within the Geminiviridae family's fourteen genera is the Begomovirus [7, 8, 9].
Begomoviruses are a genus of viruses that infect a variety of dicot plants as well as several monocot species [10]. Begomoviruses and the allied elements, known as satellites, infect economically significant plants worldwide, such as several vegetables, decorative plants, medicinal plants, fragrant plants, and major cereal crops [5, 6]. The losses caused by begomoviruses are estimated to be in the billions of US dollars, and the most important crops affected include cassava, cotton, tomato, and lady fingers, and many others [11]. In both the Old and New Worlds, begomoviruses are found in tropical and sub-tropical environments (OW, NW). These viruses have also recently spread to regions with mild climates, which may be related to global warming or other changes in the planet’s natural circumstances [11, 12]. Viruses with a single DNA component known as DNA-A (monopartite begomoviruses) or two DNA components known as DNA-A and DNA-B (bipartite begomoviruses) are the two distinct types of begomoviruses. These two components of bipartite begomoviruses are around the same size, each ranging in size from 2.5 to 3.2 kb, although each has its own distinct function, and they don't share any sequence similarities. While monopartite begomoviruses can infect plants with their DNA-A only, bipartite begomoviruses need both DNA-A and DNA-B to infect plants systematically. Begomoviruses that are monopartite are frequently linked to satellite molecules known as alpha, beta, and deltasatellites. Whitefly (Bemicia tabaci) is an insect vector that spreads begomoviruses and satellites from diseased plants to healthy plants. Begomoviruses were originally classified into two groups: the OW viruses were named monopartite begomoviruses, while the NW viruses were dubbed bipartite begomoviruses. These divisions were based on genomic organization, biogeography, and phylogeny. Whereas the NW begomoviruses are found in the western hemisphere, namely the United States of America, the OW begomoviruses are found in Asia, the eastern hemisphere, Europe, Africa, and Australia [13,14,15]. The majorities of bipartite begomoviruses are found in the Northwest and are made up of two genomic DNA components, known as DNA-A and DNA-B that are of similar sizes [16]. For crops to become seriously infected, both parts are essential [17]. DNA-A is essential for transcription and replication, while DNA-B is necessary for the bipartite begomovirus moment in plants. Bipartite begomoviruses were formerly believed to be limited to the Northwest, but a small number have recently been identified in crops in the OW. With a few notable exceptions, these bipartite begomoviruses differ genetically from those found in the Northwest [2].
To create an evolutionary tree outlining the genetic variety of viruses, phylogenetic analysis is essential. Genetic data, such as DNA or RNA sequences and protein sequences such as amino acid sequences, are the sole basis for phylogenetic analysis. This offers details on the evolutionary background of taxonomic categories. Population genetics provides support for biogeography, or phylogeography, which is based on phylogenetic understanding of biogeography. Phylogeography is the source of a significant historical perspective on public building. Inheritance and biogeography may also be used to examine in depth the lineal explanation of virus species based on the geo-climatic history of the planet. To understand the origins and diverse model of significant viral species, biogeography and phylogeny are crucial [18, 19]. Extensive research on begomovirus diseases in the two the major provinces of Pakistan (Sindh and Punjab) have been done. Nonetheless, there is currently a dearth of begomovirus research in the KP province of Pakistan. To the best of our information, there is no published data about begomoviruses infection in Swat region of KP province. Therefore, it was suggested that begomoviruses may be present in Swat region of KP and may be a risk to food security in the future.
This study aimed to explore the genetic diversity and evolution history of bipartite begomoviruses in a previously unexplored area (Swat region) of Pakistan. This will help in the monitoring of future modifications in the virus complex and may also help to devise novel control strategies against begomoviruses.
Methods![]()
Sample collection
Plant leaves showing typical begomovirus symptoms were collected from various locations of District Swat, Khyber Pakhtunkhwa province of Pakistan. Swat is also called the Switzerland of Pakistan and is famous for its greenery and tourism. The plant leaf samples were collected and stored by a standard procedure.
DNA extraction from the infected plant
A few modest modifications were made to the cetyltrimethylammonium bromide (CTAB) technique [20] in order to extract the genomic DNA from the plant leaves exhibiting begomovirus symptoms. The extracted purified genomic DNA was diluted in ultra-pure distilled water, and a 1% Agarose gel was used to verify the DNA's amount and purity.
Rolling circular amplification (RCA), cloning and sequencing of begomovirus components
By using a previously mentioned procedure [21], the RCA amplified the circular DNA of the begomovirus using the TempliPhi DNA amplification kit (GE Healthcare). The necessary reaction mixture was made and incubated for 18 to 24 hours at 28 degrees Celsius. Ultimately, the prepared mixture was treated at 65°C for 10 minutes to deactivate the activity of the Φ29 DNA polymerase enzyme. On a 1% agarose gel, the high molecular weight RCA products were verified. After restriction, endonucleases digested the RCA products into monomeric forms, a segment of approximately 2.8 kb was produced. This fragment was then cloned into the pTZ57R/T cloning vector (Fermentas) and commercially sequenced using Illumina or Sanger sequencing.
PCR Amplification, cloning and sequencing of begomovirus components
A total of 20μl PCR reaction containing 0.25U Taq polymerase, 1.6 μl MgCl2, 0.5 μM of each primer, 2 μL dNTPs, 2μL 1X Taq buffer and 1 μL DNA template was carried out in the PCR machine. Either genomic DNA or RCA product was used as a template in the PCR. The primer pair Begomo-F (5`-ACGCGTGCCGTGCTGCTGCCCCCA-3`)/ Begomo-R (5`-ACGCGTATGGGCTGYCGAAGTTSAGACG-3`) was used for the amplification of DNA-A component, while the primer pair ToLCNVB-F (5`-ACGCGTAAGGAAATCTGTGAAACAC-3`)/ ToLCNVB-R (5`-CGCGTATATTGTTTGGAGATTGGTCG-3`) was used for the amplification of DNA-B component. The reaction conditions included preheat treatment at 94℃ for 5 minutes with 35 cycles of 94℃ for 1 min, 52℃ for 1 min and 72℃ for 3 min with a final incubation of 72℃ for 10 min. The PCR amplified product was checked on 1 % Agarose gel and a size of approximately 2.8 kb was confirmed on agarose gel. The PCR amplified products were purified and cloned into a pTZ57R/T cloning vector as described above and sequenced commercially by Sanger sequencing. Primer walking and internal primers were used to complete the sequencing of entire genomic DNA components.
DNA sequence analysis, sequence retrieval and out group selection
The sequences obtained were deposited into the EMBL database and the Basic Local Alignment Search Tool (BLAST) was utilized to look for other similar sequences. Based on two and three regions, the full-length sequences of DNA-A and DNA-B were obtained independently from the National Centre for Biotechnology Information (NCBI) database. For DNA-A and DNA-B, 256 taxa with both outgroups and ingroups were included in the study. Based on the two and three sections of the bipartite begomovirus, a total of 135 taxa (124 ingroups and 11 outgroups) were taken for the DNA-A component. Similarly, 121 taxa were included for the DNA-B component based on two and three regions (115 ingroups and 6 outgroups). The outgroup for the DNA-A component was selected from the Mastrevirus species, the Maize streak virus (accession number AF329886), which taught to be diverged earlier than begomoviruses. The African cassava mosaic virus, Cotton leaf curl Multan virus, and Tomato leaf curl virus were also considered outgroups for DNA-A. For the DNA-B component, the Maize streak virus (accession number AF329889.1) was likewise considered an earlier divergence outgroup. Other viral species that were considered outgroups for DNA-B were the African cassava mosaic virus and the Bean golden yellow mosaic virus.
Estimation of time divergence and phylogenetic analysis of TLCuNDV
The DNA-A and DNA-B sequences underwent pairwise multiple alignments using Clustal X [18, 19]. The replacement rates and tree topology were determined using the BEAST software (ver. 1.7.5) and MEGA “10” programme and Jmodeltest ver. 0.1.1 were utilized for this [22, 23]. Three uncontrolled or unrestricted MCMC analyses were performed in 500,00000 steps. A sample of trees was taken every 2000 generations. Using Tracer ver.1.5, these trees were used to confirm the intersection of the immobile sampling fairness and sharing [22, 2]. Software Logcombiner (version 1.7.5) was employed to merge the log data from the three independent tests. Using the Tree Annotator programme (version 1.7.5), a tree with maximal clade credibility in relation to posterior probability (PP) was constructed from the surviving trees after the first 5000 trees were burned in [22]. The findings were visualized with Figtree version 1.4 [18, 19, 24].
Biogeographic analysis of TLCuNDV
For the potential ancestral area reconstruction of TLCuNDV [25], one event-based approach, such as S-DIVA, and two model-based methods, such as BBM [10] and DEC, were utilized. To determine the ancestral ranges of each node in S-DIVA, 25000 trees—5000 of which were discarded (burn-in) that were obtained from the MCMC output employed. Every 100 generations, samples were taken from each of the two or three maximal regions that were maintained at each node [25]. For both DEC and BBM model, the maximum clade credibility tree was used; however, for the branches lengthwise measurement the DEC model was used [18, 19]. The distribution range of TLCuNDV was split into six (6) parts or continents (from A to F). The Asia, Africa, South America, North America, Europe and Australia are represented by the alphabet A, B, C, D, E, and F, respectively.
Results![]()
Genetic diversity analysis of the full-length genome sequence of a bipartite begomovirus, TLCuNDV infecting Benincasa fistulosa plant in Swat
The entire genomic components (DNA-A and DNA- B) of a bipartite begomovirus isolate, ToLCDNV that had produced the leaf curl disease symptoms in Benincasa fistulosa (Round Gourd) plant was cloned and sequenced for the first time from a formerly unexplored area of Pakistan (Swat region). The gene bank accession numbers assigned to this bipartite begomovirus sequences were labelled as MW722701 for
Phylogenetic and biogeographic analysis of a bipartite begomovirus, TLCuNDV infecting Benincasa fistulosa plant in Swat
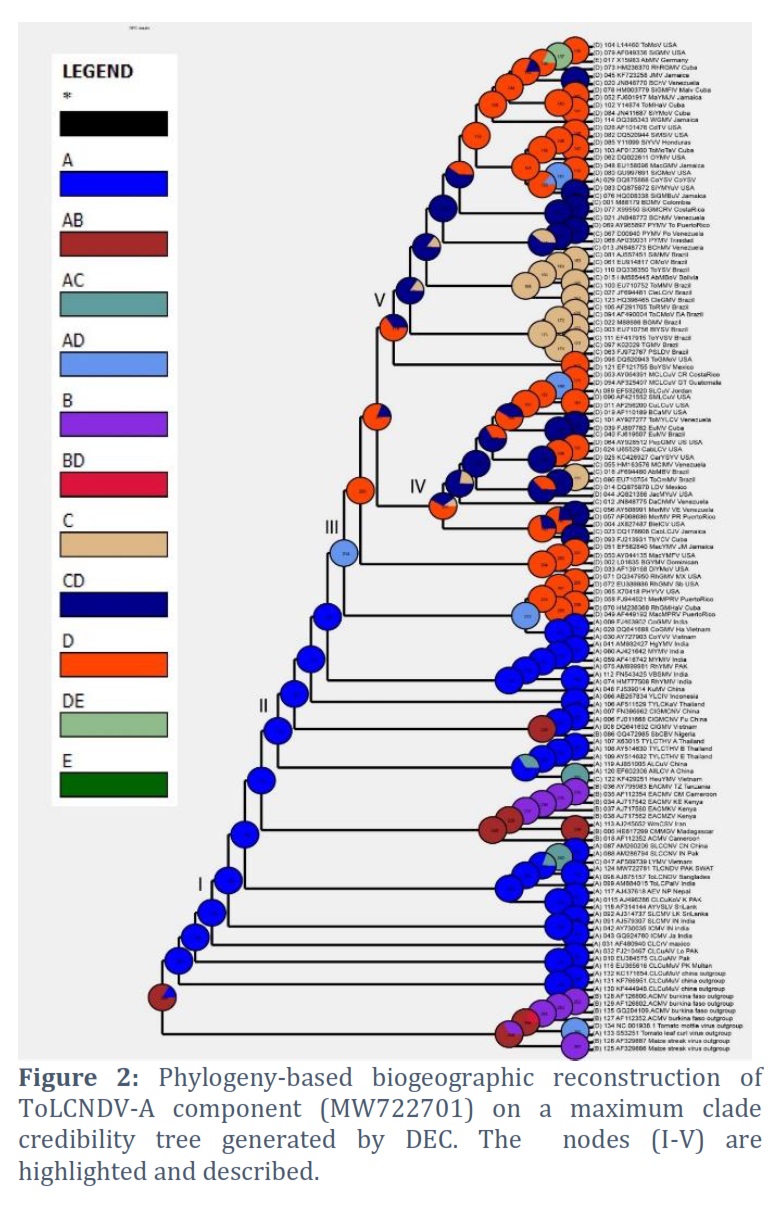
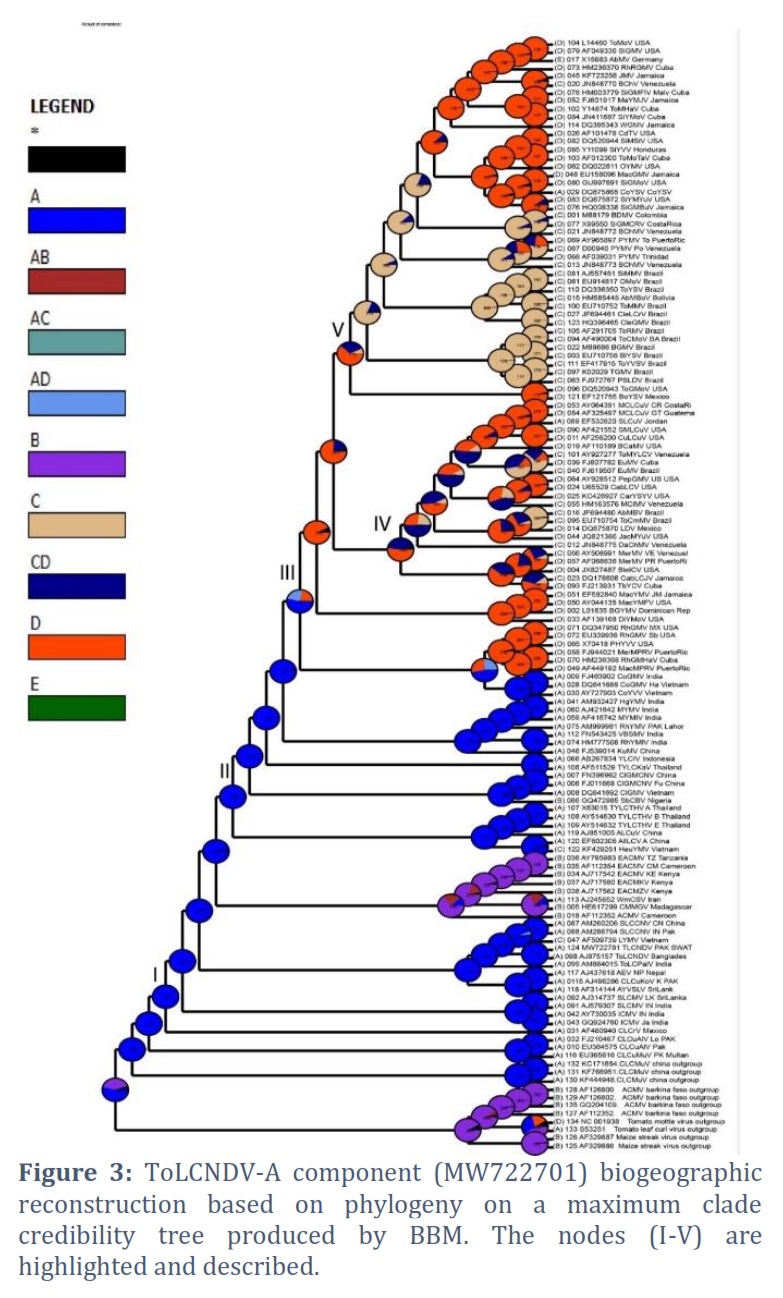
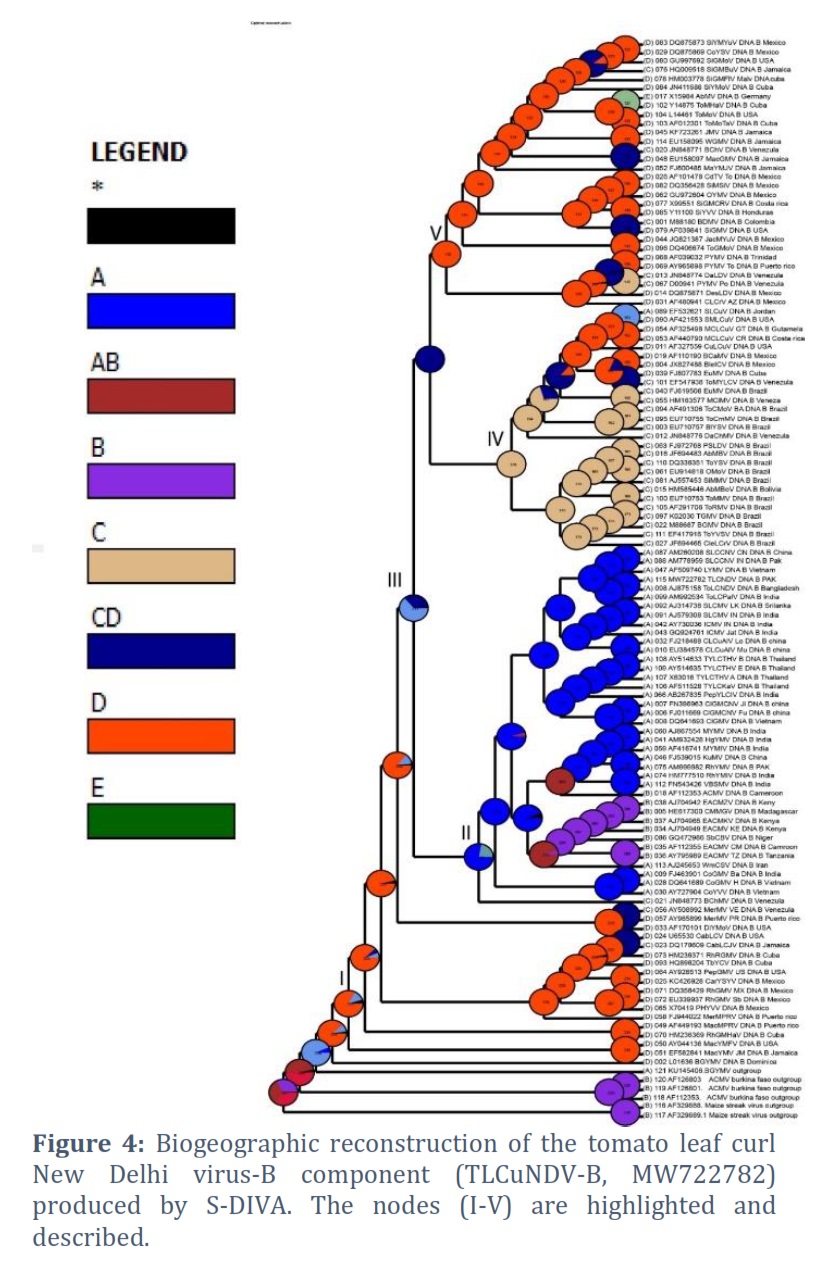
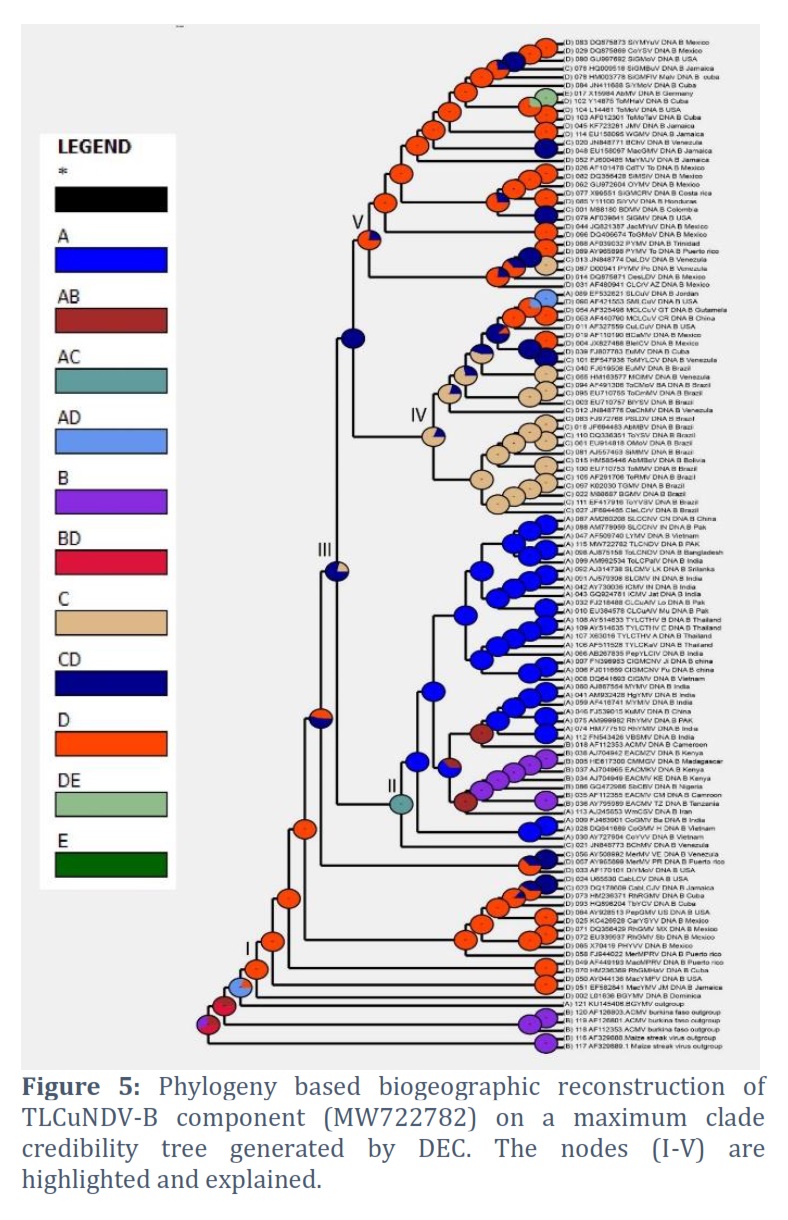
Based on posterior probability (PP) values of >95%, the fully resolved tree topologies (Fig. 1 to 5) clearly support the presence of monophyletic sub clades (nodes I–V). Phylogenetic research revealed that the DNA-A component of this virus clustered with ToLCNDV-A of Bangladesh begomovirus species and some begomoviruses species that infect cotton plant in Pakistan. Among the others are AYVSLV from Sri Lanka, SLCMV from India, Pakistan and China. Based on analysis of DNA-B component, this virus bunches with ToLCNDV-B from Bangladesh and a few other associated viruses from Thailand, Sri Lanka, Pakistan and India. The phylogenetic trees analyzed by S-DIVA, DEC, and BBM that are constructed for the DNA-A component of ToLCNDV are shown in Figs. 1, 2, and 3, respectively.
The ancestral reconstruction of ToLCNDV's DNA-A component using S-DIVA, DEC, and BBM based on two and three regions, respectively. The phylogenetic trees analyzed by S-DIVA, DEC, and BBM produced for the DNA-B component of ToLCNDV are shown in Fig. 4, and 5. The ancestral reconstruction of the DNA-B component of ToLCNDV by S-DIVA, DEC, and BBM based on two and three area, respectively.
The TLCuNDV-A (accession number MW722781) on the phylogenetic tree located at Node-I (position 124) shows that it started in India initially and made its way via China to reach Africa. In every phylogenetic tree, the results of the three studies (S-DIVA, BBM, and DEC) are comparable (Fig. 1 to 3). Position 115 of Node III displays the DNA-B of this ToLCNDV isolate (accession number MW722782), which indicates that it originated in Bangladesh and India (ToLCNDV isolate, accession number AJ875158) and expanded through China to Brazil (Fig. 4 and 5). The study also indicates that the DNA-B component developed most recently in Mexico and initially emerged in the USA via Mexico and Venezuela.
Figures & Tables

The productivity of several essential plants, such as cash crops, vegetables, aromatic and medicinal plants, some trees, and weeds, has been drastically decreased by begomoviruses [26, 27, 28]. In Pakistan’s Punjab and Sindh regions, these viruses have been thoroughly investigated, especially in cotton [29]. There are not few studies about begomoviruses infecting plants in Pakistan’s Baluchistan Province. We conducted this investigation based on the notion that begomoviruses could have spread to Pakistan’s KP region. Therefore, the Swat region of KP province was chosen for the study of begomoviruses due to its undeveloped status, mild climate, and popularity for a wide variety of vegetable crops and medicinal plants. Although begomoviruses are mostly found in tropical and subtropical regions of the world, they have lately spread to temperate regions as a result of changes in ecological conditions and international human commerce [12].
The whole genome of TLCuNDV was sequenced from a round gourd plant in Swat for the first time. The BLAST analysis shows that the DNA-A (MW722701) and DNA-B (MW722782) components of the bipartite begomovirus share over 95% of their sequence with the previously sequenced TLCuNDV from Cucurbitaceae. ToLCNDV has also been reported to be present in other cucurbits, such as Cucurbita species, Lagenaria siceraria, Benincasa hispida, and Luffa acutangula [30, 31]. This marks the initial documentation of a bipartite begomovirus from crops in Pakistan's Swat area. Other crops in the area may be seriously threatened by ToLCNDV's presence.
TLCuNDV is among the several bipartite begomoviruses that have recently been found in a number of OW crops. The same has also been noted in a number of vegetable crops grown in Punjab, Pakistan [31], as well as in Bangladesh and India [32]. Thus, we report TLCuNDV, a bipartite begomovirus that infects round gourd plants in an unexpected and undiscovered region of Pakistan.
Begomoviruses have a fairly simple structural makeup, yet their evolutionary history is quite complicated. The bipartite form of begomoviruses has been largely overlooked in evolutionary research, despite the fact that characteristics related to begomovirus-host interactions and viral evolutionary dynamics have been well investigated [33, 34]. According to some research, the evolutionary histories and selection pressures that the DNA-A and DNA-B components have encountered may differ. The conventional perception states that the satellite molecules linked to monopartite begomoviruses may have served as the ancestors of the DNA-B component of bipartite begomoviruses [16]. Many studies have been conducted to ascertain the evolutionary dynamics of begomoviruses; however, these studies have been predicated on the analysis of the bipartite begomoviruses’ DNA-A component. Because the DNA-B component of these viruses plays a significant role in the viral infectious cycle, it is imperative to take this component’s evolution into account as well [33, 34]. Here, we successfully completed a simultaneous evolutionary investigation of the bipartite begomoviruses’ DNA-B and DNA-A components. To reconstruct and arrange the evolution of the DNA-A and DNA-B components of ToLCNDV, as well as to identify their ancestral locations, phylogenetic and biogeographic studies were conducted. Which of the bipartite and monopartite begomoviruses emerged first, and where they could have originated, is still up for debate? It is believed that they could have co-evolved [16, 35]. A general grand scheme of the ToLCNDV's expansion was proposed by this study.
Since none of the New World begomoviruses carry the AV2 gene, it is likely that they all descended from a single parent. Furthermore, the genomes of begomoviruses from the OW and NW differ relatively little from one another. Therefore, it is suggested that despite their varied geographic distribution, bipartite and monopartite begomoviruses may have developed from a single parent. There is no chronological relationship between the begomoviruses and the satellites they are associated with, and the satellites would not function without the begomoviruses. Since begomoviruses had to evolve before satellites, it is possible that these two different types of molecules came from different parents. Furthermore, it's feasible that the OW's monopartite begomoviruses originated before the NW's bipartite begomoviruses [2]. It is suggested that begomoviruses could have started in the OW before moving on to the NW. The divergence of bipartite begomoviruses is believed to have occurred in two of the eight areas proposed for begomovirus expansion or divergence (South and North Americas); the remaining six areas (Indian subcontinent, China, Europe, Australia, Japan and Africa) are thought to have caused the divergence of monopartite begomoviruses and their satellites [35]. Phylogenetic analysis revealed that within the distinct trees produced by the three separate analyses, the virus isolate that was infecting Benincasa fistulosa plants in Swat, ToLCNDV-A component (MW722701), and the plants that were infected by CLCuMuV, CLCuKoV, and CLCuAV that were isolated from Pakistan, clustered together (at node 1). SLCM, AYVSLV, and ToLCPaV from Sri Lanka, SLCCNV from China and Pakistan, ICMV, SLCM and ToLCPaV from India are among the other begomoviruses grouped together in the same group (Fig. 1 to 3). ToLCNDV-B isolates from Bangladesh and other virus isolates from India, Thailand, Pakistan, and Sri Lanka are clustered together (at node III within the various trees constructed by three independent studies), according to the analysis of the ToLCNDV-B component (MW722782) (Fig. 4). The results showed that it started in India, spread via China, and eventually reached in Africa. Phylogenetic based research suggests that the DNA-B component of begomoviruses evolved in Mexico more recently, after having first originated in the USA via Mexico and Venezuela. More than forty distinct plant species from the Mediterranean, Far East and Middle East area of Asia have previously been reported to be infected with ToLCNDV [31]. This validates ToLNCDV's extraordinary efficacy as a worldwide pathogen. In particular, we are reporting a second pathogen host shift caused by the same strain of ToLCNDV, which affects several other plant host species belonging to the families Solanaceae, Fabaceae, Euphorbiaceae, Cucurbitaceae, and Malvaceae. Begomoviruses and their associated satellites rely solely on Bemicia tabaci as their propagation vector. Thus, Bemicia tabaci could have played a significant role in the begomovirus complex's evolution and divergence [34]. Furthermore, wind and trade in infected goods may have a major role in the global spread of begomoviruses. Even yet, it is challenging to determine the precise start of begomovirus evolution since no fossil traces of the viruses exist at this point in time. It’s possible that the oceans, mountains, and deserts found in many different parts of the world and regions prevented these viruses from spreading farther [2]. Using cutting-edge sequencing techniques, additional viral isolates from the various study sites and plant hosts must be sequenced. Furthermore, the use of contemporary bioinformatics methods might lead to a more precise analysis of the viral sequences.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research, King Saud University, for funding through the Vice Deanship of Scientific Research Chairs, the chair of date palm research. We also highly acknowledge Higher Education of Pakistan for providing funding for this project under National Research Program for Universities (NRPU) Project No. 6835/KPK/NRPU/R&D/HEC/2016.
Conflict of Interest
The authors declare that there is no conflict of interest.
FA and RSK gave the basic idea. CHA and ZAH wrote the first draft of the manuscript. CHA conducted the experiments. KH, MAR, HS and AUD analyzed the data. MAAS writes the final draft.
![]() References
References
- Gutierrez C. Geminivirus DNA replication. Cellular and Molecular Life Sciences, (1999); 56: 313–329.
- Ha. C, Coombs S, Revill P, Harding R, Vu M, Dale J.A. Molecular characterization of begomoviruses and DNA satellites from Vietnam: additional evidence that the NW geminiviruses were present in the OW prior to continental separation. Journal of General Virology, (2008); 89: 312–326.
- Yu Y, Harris AJ, Xingjian H. RASP version 2.1b. 2011, Available at http:/mnh.scu.edu.cn/soft/blog/RASP.
- Brown G. Public participation GIS (PPGIS) for regional and environmental planning: Reflections on a decade of empirical research. Journal Of the Urban & Regional Information Systems Association, (2012); 24(2).
- Uniyal, A P, Mansotra K, Yadav S K, Kumar V. An overview of designing and selection of sgRNAs for precise genome editing by the CRISPR-Cas9 system in plants. 3 Biotech, (2019); 9(6): 1-19.
- Varsani, A, Roumagnac P, Fuchs M, Navas-Castillo J, Moriones E, Idris A, Martin D. P. Capulavirus and Grablovirus: two new genera in the family Geminiviridae. Archives of Virology, (2017); 162 (6): 1819-1831.
- Roumagnac, P, Granier M, Bernardo P, Deshoux M, Ferdinand R, Galzi S, Mesléard F. Alfalfa leaf curl virus: An aphid-transmitted geminivirus. Journal of Virology, (2015);89(18):9683-8.
- Roumagnac, P, Lett J. M.; Fiallo-Olivé E, Navas-Castillo J, Zerbini F. M.; Martin D. P.; Varsani A. Establishment of five new genera in the family Geminiviridae: Citlodavirus, Maldovirus, Mulcrilevirus, Opunvirus, and Topilevirus. Archives of virology, (2021); 1-16.
- Varsani, A, Roumagnac P, Fuchs M, Navas-Castillo J, Moriones E, Idris A, Martin D. P. Capulavirus and Grablovirus: two new genera in the family Geminiviridae. Archives of Virology, (2017); 162 (6): 1819-1831.
- Zerbini, F M, Briddon R W, Idris A, Martin D P, Moriones E, Navas-Castillo J, Consortium I.R. ICTV virus taxonomy profile: Geminiviridae. Journal of General Virology, (2017); 98 (2): 131.
- Mansoor, S, Zafar Y, Briddon, R. W. Geminivirus disease complexes: the threat is spreading. Trends in plant science, (2006); 11(5): 209-212.
- Navas-Castillo, J, Fiallo-Olivé E, Sánchez-Campos S. Emerging virus diseases transmitted by whiteflies. Annual Review of Phytopathology, (2011); 49: 219-248.
- Padidam, M, Sawyer S, Fauquet C. M. Possible emergence of new geminiviruses by frequent recombination. Virology, (1999); 265(2): 218-225.
- Paximadis, M, Idris A. M.; Torres-Jerez, I, Villarreal A, Rey M. E. C.; Brown J. K. Characterization of tobacco geminiviruses in the Old and New World. Archives of Virology, (1999);144 (4): 703-717.
- Rybicki E. P. A phylogenetic and evolutionary justification for three genera of Geminiviridae. Archives of Virology, (1994); 139 (1-2): 49-77.
- Briddon, R. W . Effects of genetic changes to the begomovirus/betasatellite complex causing cotton leaf curl disease in South Asia post-resistance breaking. Virus Res. (2014);186: 114–119
- Rojas, M R, Hagen C, Lucas W J, Gilbertson R L. Exploiting chinks in the plant's armor: Evolution and emergence of geminiviruses. Annual Review of Phytopathology, (2005); 43: 361-394
- Ali, S S, Pfosser M, Wetschnig W, Martínez‐Azorín M, Crespo M.B, Yu Y. Out of Africa: Miocene Dispersal, Vicariance, and Extinction within Hyacinthaceae Subfamily Urgineoideae. Journal of Integrative Plant Biology, (2013); 55 (10): 950–964.
- Ali, S S, Yan Yu, Martin P, Wetschnig W. Inferences of biogeographical histories within subfamily Hyacinthoideae using S-DIVA and Bayesian binary MCMC analysis implemented in RASP (Reconstruct Ancestral State in Phylogenies). Annals of Botany, (2012); 109 (1): 95–107.
- Rehman S U, Muhammad K, Sher H, Que Y, Ali R, Ali S, Hassan I, Rahat MA. Molecular Analysis of Cold Responsive (COR) Genes in Selected Sugarcane and Saccharum spontaneum L. Advancements in Life Sciences. (2023) 21;9(4):547-51.
- Habile D, Kober S, Jeske H. Rolling circle amplification revolutionizes diagnosis and genomics of geminiviruses. Virol Methods. (2006); 135 (1): 9-16.
- Drummond A J, Suchard M. A, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, (2012); 29 (8): 1969-1973.
- Posada D. j ModelTest: phylogenetic model averaging. Molecular Biology and Evolution, 2008; 25 (7): 1253-1256.
- Rocha C S, Castillo-Urquiza G P, Lima A T, Silva F N, Xavier C. A, Hora-Júnior B. T, Zerbini F M. Brazilian begomovirus populations are highly recombinant, rapidly evolving, and segregated based on geographical location. Journal of virology, (2013); 87(10): 5784-5799.
- Moyle RG. Andersen MJ, Oliveros CH, Steinheimer FD, Reddy SD. Phylogeny and biogeographic of the core babblers (Aves: Timaliidae). Systematic Biology, (2012); 61: 631-651.
- Saeed ST, Samad A. Emerging threat of begomoviruses to the cultivation of medicinal and aromatic crops and their management strategies. Virus Disease (2017); 28(1): 1-17.
- Uniyal, AP, Mansotra K, Yadav S. K, Kumar V. An overview of designing and selection of sgRNAs for precise genome editing by the CRISPR-Cas9 system in plants. 3 Biotech, (2019); 9(6): 1-19.
- Inoue-Nagata, AK, Bassanezi R B, Belasque J, Amorim L, Macedo MA, Barbosa JC, Savary S. The importance of primary inoculum and area-wide disease management to crop health and food security. Food Security, (2016); 8(1): 221-238.
- Mansoor S, Amin I, Briddon RW. Geminiviral diseases of cotton. Stress Physiology in Cotton. 2011:125.
- Moriones E, Praveen S, Chakraborty S. Tomato leaf curl New Delhi virus: an emerging virus complex threatening vegetable and fiber crops. Viruses. (2017); 9 (10): 264.
- Ali Z, Ali S, Tashkandi M, Zaidi SS, Mahfouz MM. CRISPR/Cas9-mediated immunity to geminiviruses: differential interference and evasion. Scientific reports. (2016); 6(1):26912.
- Sahu AK, Sanan-Mishra N. Complete genome sequence of a new bipartite begomovirus associated with leaf curl disease of Capsicum annum. 3 Biotech (2020); 10 (5):235.
- Rodríguez-Negrete EA, Morales-Aguilar JJ, Domínguez-Duran G, Torres-Devora G, Camacho-Beltrán E, Leyva-López NE, Voloudakis AE, Bejarano ER, Méndez-Lozano J. High-throughput sequencing reveals differential begomovirus species diversity in non-cultivated plants in Northern-Pacific Mexico. Viruses. (2019);11(7):594.
- Srivastava A, Pandey V, Al-Sadi AM, Shahid MS, Gaur RK. An insight into emerging begomoviruses and their satellite complex causing papaya leaf curl disease. Current Genomics. (2023);24(1):2.
- Nawaz-ul-Rehman MS, Fauquet CM. Evolution of geminiviruses and their satellites. FEBS letters. (2009); 583 (12):1825-32.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0