

Full Length Research Article
Acquiring insights through a sequence-based approach to the critical Zika virus MTase domain
Banan Atwah1, Saad Alghamdi1, Nizar H. Saeedi2, Abdulrahman Alzahrani3, Rashed Mohammed Alghamdi4, Asif Hussain Akber5, Mohammed Yahya Al Qahtani5, Atiah Abkar Yahya Mujarribi5, Saeed Saleh Al Qahtani6, Ahmed Mohammed Faqihi6, Mohammad Azhar Kamal7*
Adv. life sci., vol. 11, no. 2, pp. 430-437, May 2024
*– Corresponding Author: Mohammad Azhar Kamal (ma.kamal@psau.edu.sa)
Authors' Affiliations
2. Department of Medical Laboratory Technology, Faculty of Applied Medical Sciences, University of Tabuk, Tabuk – Kingdom of Saudi Arabia
3. Department of Applied Medical Sciences, Applied College, Al-Baha University, Al-Baha City – Kingdom of Saudi Arabia
4. Department of Laboratory Medicine, Faculty of Applied College, Al-Baha University – Kingdom of Saudi Arabia
5. Central Military Laboratory and Blood Bank Department – Virology Division, Prince Sultan Military Medical City, Riyadh – Kingdom of Saudi Arabia
6. Central Military Laboratory and Blood Bank Department – Microbiology Division, Prince Sultan Military Medical City, Riyadh – Kingdom of Saudi Arabia
7. Department of Pharmaceutics, College of Pharmacy, Prince Sattam Bin Abdulaziz University, Alkharj – Kingdom of Saudi Arabia
[Date Received: 31/08/2023; Date Revised: 16/01/2024; Date Available Online: 18/04/2024]
Editorial Note on Version of Record
31 May 2025: This article has been corrected. See https://doi.org/10.62940/als.v13i0.4274 for more information.
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: ZIKV is one of the re-emerging arboviruses (viruses carried by arthropods), which is spread through the Aedes mosquito. It is an RNA virus with only one strand that is appropriate to the family Flaviviridae’s Flavivirus (genus) & has been linked to other Flaviviruses such as the West Nile virus, chikungunya virus, & dengue (DENV) virus. The envelope, precursor membrane, and capsid are three structural proteins, and seven nonstructural proteins are also encoded by the Zika virus genome.
Methods: We conducted an in-silico analysis of the Zika virus’ MTase domain protein for this publication. We predicted that methylation would play a significant role in the available Prosite, Pfam, and InterProScan tools to aid in locating the MTase domain. Along with alignment, amino acid composition of, charged amino acids, atomic level studies, & molecular weight, we also make predictions for these variables, including theoretical Pi.
Results: We also examine the MTase domain’s simulated structure (alpha helix, beta sheet, turn) and its specifics, including secondary structure. We also pinpoint the locations where proteins, DNA, and RNA bind. Potential phosphorylation sites can be found on the Ser, Thr, and Tyr residues in the MTase domain.
Conclusion: These outcomes imply a complicated interaction between different phosphorylation modifications that modulates the activity of the MTase domain. To fully appreciate the auxiliary and practical perspectives and to clarify the varied roles of PTM in the MTase domain will be a primary goal of future study.
Keywords: I-TASSER; Secondary structure; Prosite; α-helix; Pfam; InterProScan; Binding sites; Posttranslational modification; SOPMA; Phyre2
Introduction![]()
Several rising & re-emerging infections have taken a significant toll on global public health [1]. Recently reported diseases include swine influenza, severe acute syndrome (respiratory), Middle East respiratory syndrome, Ebola virus disease, & Zika virus (ZIKV) infection [2]. The most notable of these infections is the ZIKV infection, which has crossed all international borders and has been recorded from all corners of the globe [3,4]. The Zika virus is one of the members of the Flaviviridae virus family. It is carried primarily through Aedes mosquitos that are prevalent for the duration of the day, including Aedes aegypti & Aedes albopictus [5]. It has been recognized to take place along a limited zone stretching from Africa to Asia since the 1950s. Subsequently 2007 to 2016, the virus traveled eastward over the Pacific Ocean to the Americas, resulting in the Zika virus epidemic from the period 2015 to 2016 [6, 7]. ZIKV is a small +ve sense RNA virus with an envelope. [8]. Its genome generates a polyprotein (about 3423 amino acids long) composed of seven proteins that are nonstructural NS-1, NS-2A, NS-2B, NS-3, NS-4A, NS-4B, & NS-5 & three proteins which are structural capsid, membrane, & envelope [9]. Having 904 amino acids, NS5 has the most significant number of nonstructural proteins [10, 11]. It consists of two domains: an N-terminal region and C-terminal region. The Methyltransferase (MTase) domain belongs to the N & RNA-dependent RNA polymerase (RdRp) to the C-terminal region [12]. The NS5 & RdRp domain helps facilitate viral RNA synthesis via a de novo beginning strategy [13]. The NS5 Methyltransferase domain methylates the RNA cap using SAM (S-adenosyl-L-methionine as the methyl) donor to create N-7-methyl-guanosine & 2′-O-methyl-adenosine [14]. In this publication, we examine the composition of of the amino acid-based sequencing analysis. We modeled the MTase domain’s structure and validated it using Ramachandran plots, which included details on the secondary structure, including the α-helix (alpha helix), β-sheet (beta sheet), β turns (beta-turn), & random coil. We also identified protein, DNA, & RNA binding sites in the MTase domain, which comprises potential phosphorylation sites on Ser, Thr, & Tyr residues. These results imply a complicated interaction between different phosphorylation modifications that modulates the activity of the MTase domain. To fully utilize techniques from the MTase area will be a crucial goal of future research.
Methods![]()
The sequence of amino acids and their composition
The Zika virus MTase domain amino acid sequence (263 amino acids) was sent to ExPASy Server via the link (https://web.expasy.org/protparam/), which created an aligned amino acid pattern with the number of amino acids (aa), molecular weight (mw), & theoretical PI [15,16]. The whole amino acid content of the protein (CSV format) is entered into an excel spread sheet, & a good depiction of numerous residues with percentages covered throughout the protein is drawn [17].
I-ITASSER predicts three-dimensional structure
The Methyl transferase domain (MTase) sequence of 263 amino acids was obtained in FASTA format from the database (Swiss Prot). The 3D model was created with the I-TASSER service & creates a model (3D) of the request sequence [18]. This server was chosen because of its availability, blended modelling approach, & performance in the CASP cooperation. The I-TASSER technique contains general steps such as threading, structural assemblage, model assortment, refining, & structure founded functional annotation [19, 20]. The request sequence was then LOMETS [21] routed through the representative PDB structure collection. The eminence of template alignment was verified using Z-score, & the finest one was employed for additional evaluation. The incessant fragments are then processed in the following step. The subsequent stage eliminated the straight pieces in threading alignments from a collected structure model of aligned sections. Unaligned parts have low modelling accuracy while incorporating aligned regions offer high precision. Therefore, these template pieces must remain stiff during the simulation phase to achieve a high-resolution structure. Cα/side chain correspondence, H-bonds, hydrophobicity, spatial restrictions from threading templates & sequence-based connection expectations from SVMSEQ [22, 23] are all included in the simulation. SPICKER [24] was used to cluster the conformations created throughout the refinement simulation process, & the usual of the 3D bring together of all the gathered structures was determined to be obtained. The carefully chosen cluster centroids were employed once more during the refining phase. TM-align was used to identify the PDB structures operationally near the cluster centroids [25]. The finishing structural models were created through REMO [26], which used cluster centroids from the second-round simulation as contribution. Finally, the purpose of a 3D model of the request protein was anticipated through comparing it to proteins with notorious structure & function in the PDB. The purposeful analogs were graded according to their TM score, RMSD, sequence identity, & structural alignment coverage. The C-score (confidence score) measured the anticipated model’s quality, which varied from -5 to 2.
Prediction model validation
The Ramachandran plot verified the finest model of the MTase domain expected by I-TASSER even more. The anticipated model’s conformation was derived by way of evaluating the phi (Φ) psi (Ψ) torsion angles with the available tool (PROCHECK) web service. PROCHECK’s Ramachandran plot terms an excellent model by means of over 90% residues in the maximum favored area [27].
The secondary structure extrapolation
Utilizing the online accessible server for phyre 2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index), the secondary structure of the MTase domain (263 amino acids) of the Zika virus was predicted. The fact that the MTase domain may accommodate strands such as -helix, beta strand, and disordered is significant [28].
SOPMA is used to analyze a protein’s secondary structure
In this exercise, we apply SOPMA (Self-Optimized Prediction method with Alignment) to analyze the secondary structure of a protein (MTase domain). A protein’s function is significantly influenced by its shape. A protein must bind to other molecules in a specific way to function correctly. Every protein has a unique structure as a result. Primary, secondary, tertiary, & quaternary protein structures are categorized. The proteins’ secondary, tertiary, & quaternary structures are molded after they fold from their primary sequences during protein synthesis. SOPMA is a technique for envisaging a protein’s secondary structure from its primary sequence [29].
Envisaging the binding site
The protein’s primary sequence is extracted from the NCBI database and transmitted to the server (http://www.predictprotein.org), creating numerous sections of binding sites, including protein, DNA, & RNA binding sites. In this viewer, the various coloured boxes stand in for anticipated characteristics that pertain to particular positions in the crucial sequence. All fundamental biological processes, including mechanisms, depend on proteins, DNA, and RNA [30].
Posttranslational modifications analysis
Post-translational modifications (PTMs) occur on nearly all proteins studied to date. Expanded knowledge of the potential PTMs of an objective protein may improve our understanding of the sub-atomic processes in which it participates [31]. These changes frequently significantly impact the functionality of an adjusted protein. The MTase domain protein’s sequence was found and entered into the Simple Modular Architecture Research Tool, along with PTMs and any anticipated affiliations that might be relevant. The unit sequence database was used to retrieve the sequence information needed to expect the phosphorylation & modification sites in the MTase (Zika virus) protein. NetphosK 1.0 (http://www.cbs.dtu.dk/services/NetPhosK/) was with to predict the ability of kinases designed to phosphorylate Thr Ser, & Tyr in the MTase domain of the Zika virus [31].
Results![]()
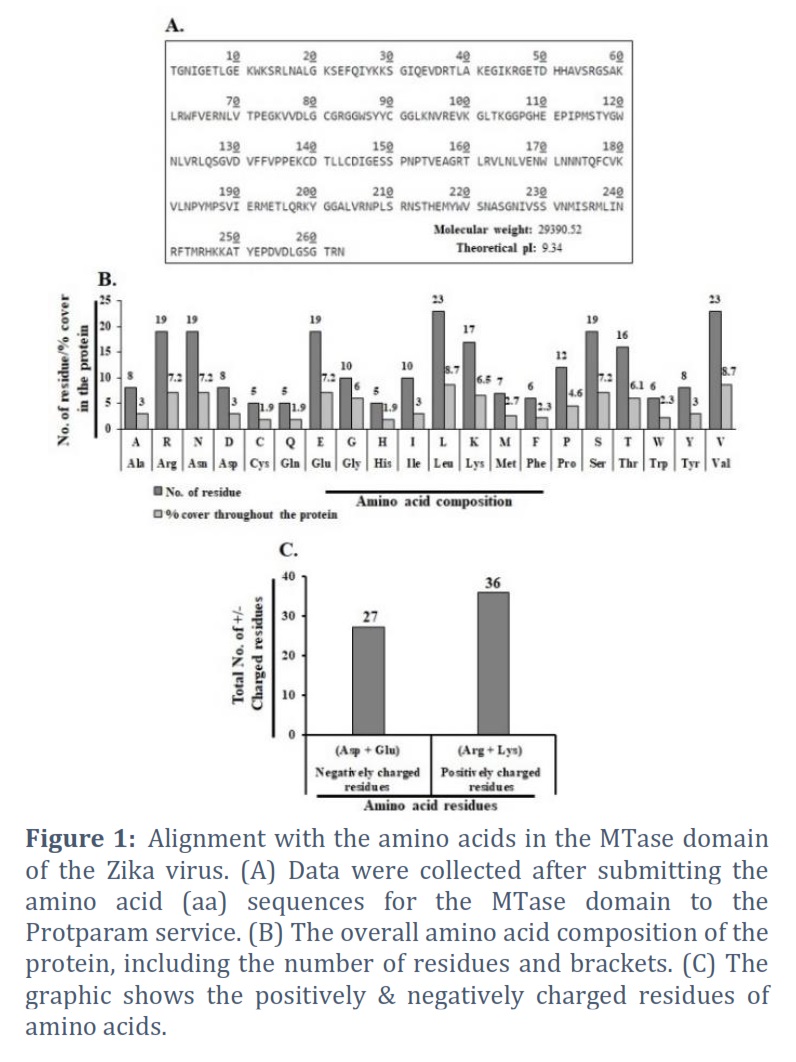
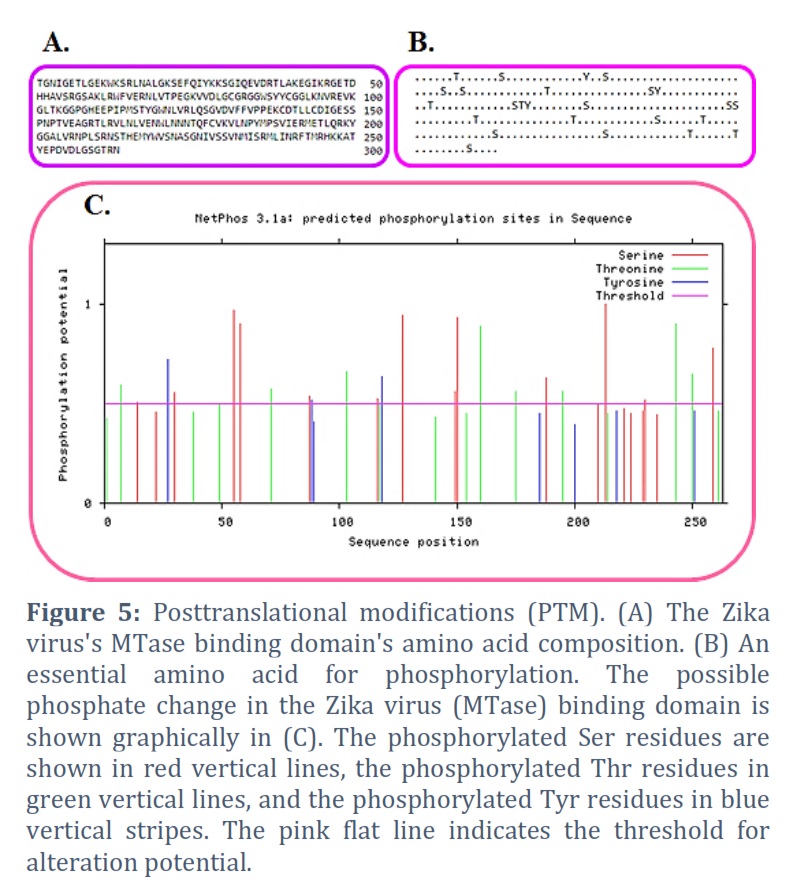
An amino acid (263) Zika virus binding domain (MTase) sequence. Utilizing the Protparam server, the amino acid sequence alignment was completed. The molecular mass (29390.52) and theoretical Pi (9.34) were discovered (Figure 1A). The total amount of amino acids in the protein as well as its paternity and residue counts are shown in Figure 1B. A:3.2, R:7.2, N:7.2, C:1.9, Q:1.9, E:7.2, G:6.0, H:1.9, I:3.0, L:8.7, K:6.5, M:2.7, F:2.3, P:4.6, S:7.2, T:6.1, W:2.3, Y:3.0, and V:8.7 are the amino acid residues that composition of the majority of the protein, respectively. We also estimated the positively and negatively charged residues in this protein. In Figure 1C, we discovered that there are +ve charged amino acids (Arg + Lys) in number 36 & correspondingly, -ve charged amino acids (Asp + Glu) in number 27. We have calculated the instability index (37.79) and aliphatic index (77.34).
Prediction of three-dimensional structures
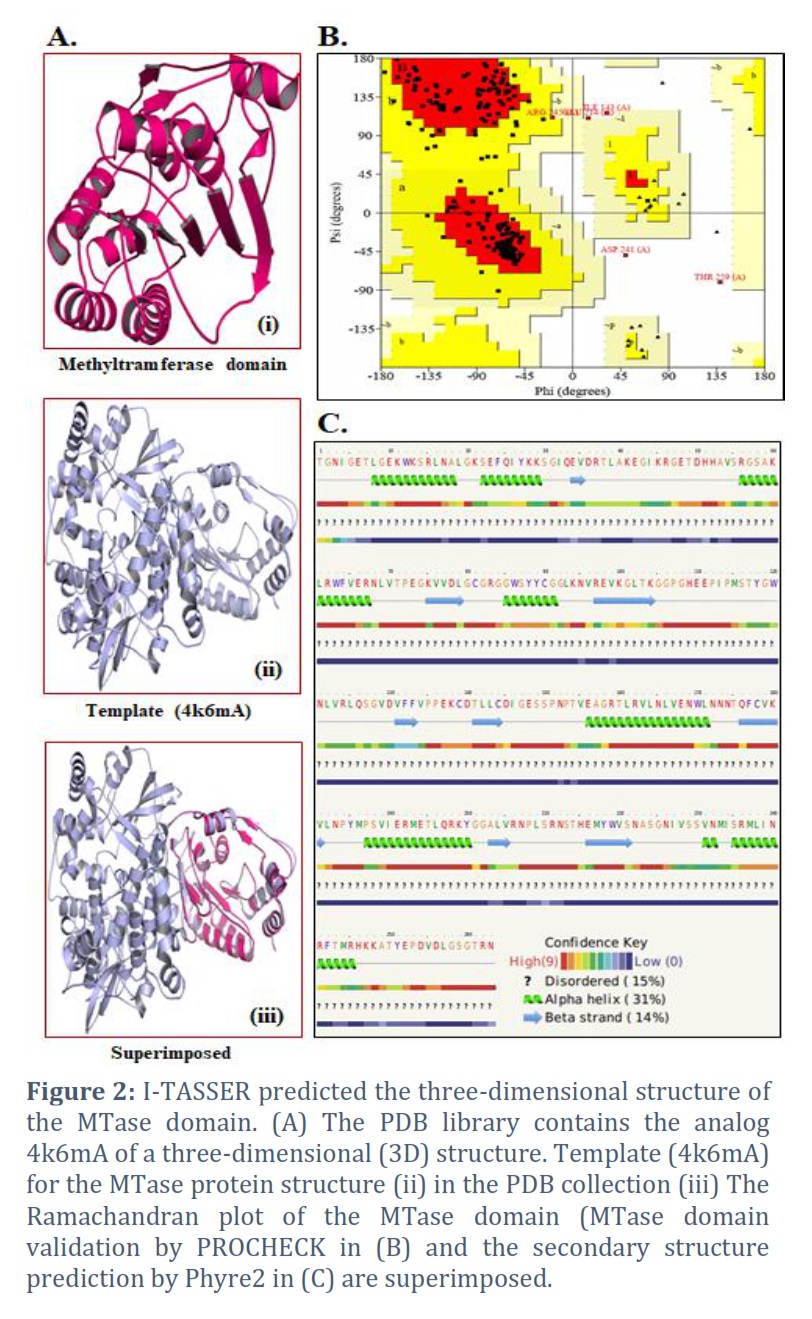
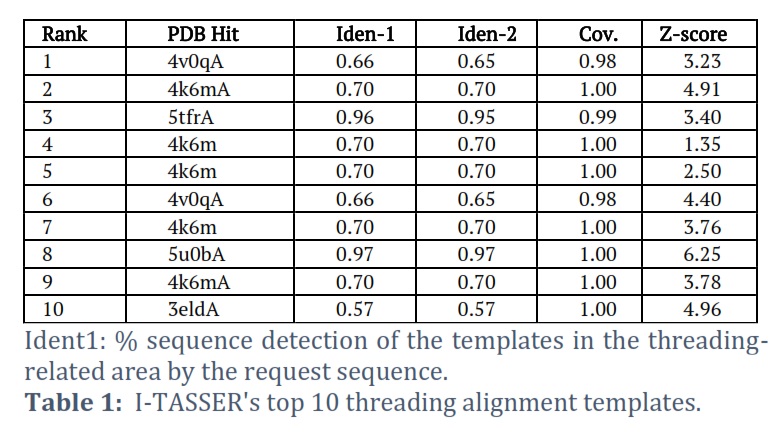
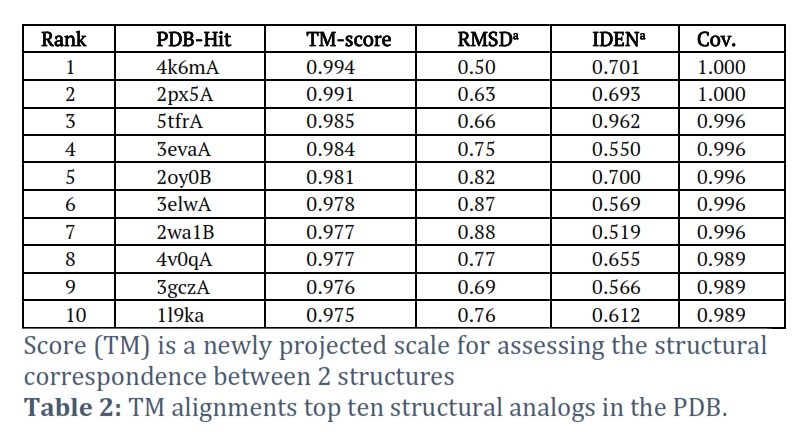
I-TASSER was able to effectively obtain the projected model of the MTase domain protein & its 3D (three-dimensional) coordinate file in PDB format. A anticipated secondary structure by way of a confidence score (range between 0 & 9), an indication of solvent convenience, five anticipated structures by means of a C-score, the top ten PDB templates applied in arrangement, the top 10 PDB structural, functional protein (analogs), & binding site residues are among the results acquired from the server. The model through the premier C-score & lowest RMSD was determined to be the Model MTase domain (Figure 2A). The LOMETS meta-server determined the top ten threading templates for the request protein sequence’s MTase domain (Table 1). Normalized Z-score is typically used to estimate threading alignment. However, a normalized Z-score greater than one (Z-score >1) value indicates an exacting alignment. Still, it does not provide a meaningful indicator of modeling accuracy in the case of a slight alignment of a lengthy query sequence. Suitable homology is defined as the proportion of sequence identity in the threading aligned area (Iden1) & the entire chain (Iden2) (Table 1). PDB library’s 4k6mA, which has a TM-score of 0.994, was the finest structural analog of the top-scoring I-TASSER model through the structural alignment platform TM-align (Table 2).
Ident2: the percentage of the complete template chain sequences the request sequence recognizes .Cov, which stands for threading alignment reporting, is the number of aligned residues divided by the request protein’s length. The threading alignments’ regularized Z-score is known as the Z-score. A noble alignment is indicated by an alignment with a standardized Z-score >1 and vice versa.
RMSDa is the RMSD between structurally aligned residues determined by TM-Align. IDENa represents the percentage of sequence uniqueness in the structurally aligned region. Cov denotes the TM alignment coverage and is equal to the number of structurally aligned residues that are aligned along the length of the protein.
A review of the secondary structure and the projected model
The finest predictable model’s Ramachandran plots were received as of PROCHECK servers & demonstrated the model’s dependability. A high-quality model of the MTase domain can be seen in the PROCHECK Ramachandran plot, which displayed 97.8% residues in highly favored regions & 0.5 percent residues in further allowable regions, for a total of 98.3% residues in the allowed areas (Figure 2B). The strands, -helix, and beta-strand in the MTase domain were evaluated by secondary-structure prediction methods (Figure 2C).
SOPMA-based protein analysis
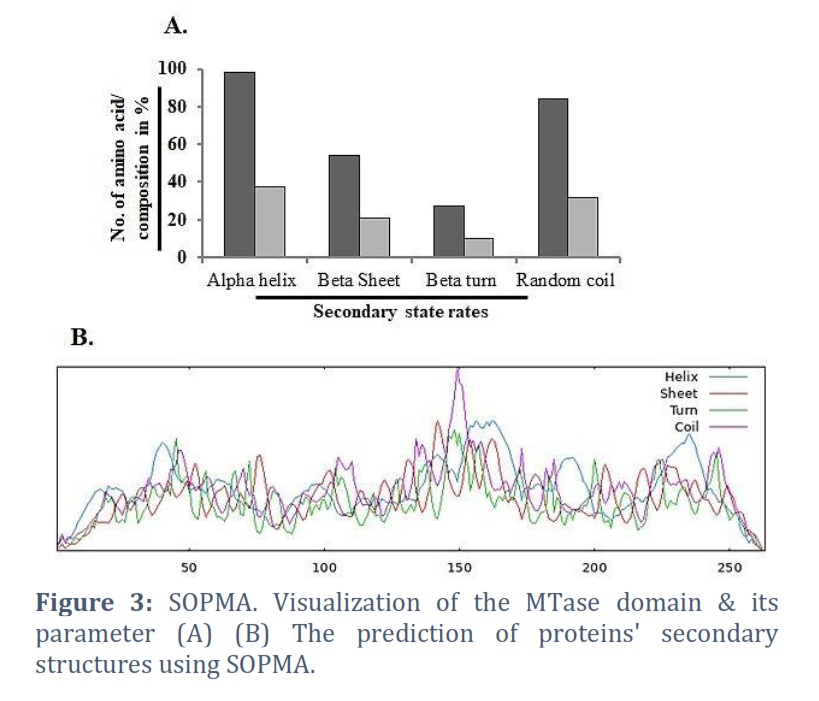
A tool for predicting the secondary structure of a protein is the Self-Optimized Prediction method with Alignment (SOPMA) [30, 31]. The secondary structure of proteins may now be predicted more successfully thanks to a novel technique called SOPMA. α-helix, turn, random coil & β-sheet, all forecast amino acids gave a three-state portrayal of the secondary structure using the enhanced SOPM technique (SOPMA). The amino acid composition of of the alpha helix is 101, the extended strand is 54, the beta turn is 22, and the random coil is 86 (Figures 5A & 5B).
Extrapolation of the binding location
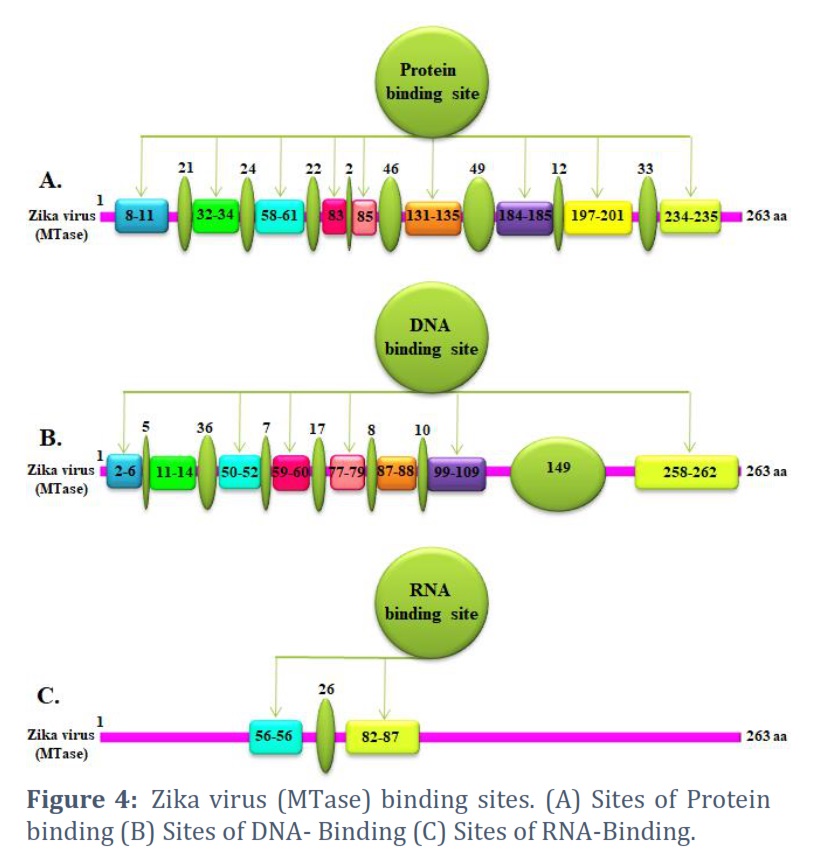
The core sequence (protein) is retrieved from the NCBI database & submitted to the server (http://www.predictprotein.org) to make several sections of binding sites, such as protein, DNA, & RNA binding sites. Figure 4A depicts the schematic for the protein binding site. The binding sites have eight locations with varied amounts of amino acids. It is also acceptable to place required ligands or interacting proteins in this crucial position. Figure 4B shows the nine locations of the DNA binding site. This interaction with the RNA binding site in Figure 4C may help the process at the DNA level. This belongs to the group of proteins called RNA helicases.
It will also be beneficial in terms of biological support. This observer displays predicted types corresponding to locations in the principal sequence using the various colored boxes. All fundamental biological processes, including mechanisms, are strongly influenced by proteins, DNA, and RNA.
Posttranslational modifications
Protein post-translational modifications (PTMs) inside & between relating proteins constitute a source of known and hypothesized functional connections [32, 33, 34, 35, 36]. It presently has a sequence of 263 amino acids (Figure 5A). The 25 counts of amino acids that composition of the 263 amino acids include the following phosphorylation sites: T, S, Y, S, S, S, T, S, Y, T, S, T, Y, S, S, S, T, T, S, S, T, T, and S (Figure 5B). The sorts of PTMs and their number are displayed on the graph. Figure 5C shows the predictions made for the Ser (Serine), Thr (Threonine), and Tyr (Tyrosine) amino acids for the MTase domain of the Zika virus using Netphos 2.0. The results of NetphosK 1.0 illustrate that MTase is the primary possible kinase to adapt these sites and can predict the ability of kinases linked to the MTase domain autophosphorylation locations.
Figures & Tables
The Flavivirus-Zika virus (ZIKV), declared a public health emergency by the World Health Organization (WHO) on February 1, 2016. ZIKV is a virus from the Flaviviridae family and Aedes mosquitoes act as vectors [37-38]. The NS5 protein, which has residues and two subunits necessary for replication, is the largest protein encoded in the viral genome. The N-terminal residues are occupied by methyltransferase (MTase), which modifies the viral RNA [39]. According to functional characterization, the N-7 and 2′-O-methyltransferase activities of ZIKV MTase help in the formation of a cap-1 structure on the viral mRNA. The interface between the MTase and RdRp domains has been reported to be critical for the regulation of dengue virus (36-38) or Japanese encephalitis virus [40]. Furthermore, dengue virus RdRp has been shown to efficiently initiate and prolong RNA polymerization via the MTase domain [41]. Given that the ZIKV MTase shares a high degree of structural and functional similarity with other flavivirus MTases, research on the DENV MTase may be helpful in the creation of antiviral drugs that specifically target the ZIKV MTase. We examined three classes of DENV MTase inhibitors—cap analogs, SAM analogs, and allosteric inhibitors for ZIKV MTase—in order to further evaluate this. According to our findings, these molecules can be used as a basis for MTase inhibitor identification. As anticipated, ZIKV MTase is similarly inhibited by Cap and SAM analogs, which have been previously reported to inhibit viral MTases. Compound selectivity is still the most significant obstacle when aiming to target the SAM binding site in order to prevent cellular MTase inhibition. In this sense, the structure of the ZIKV MTase indicates that, similar to the DENV MTase, an increase in specificity may be obtained by utilizing the conserved hydrophobic cavity next to the SAM binding site [42]. In conclusion, we offer here the first analysis of ZIKV MTase’s structure and function. This study shows how similar the structures of the DENV and ZIKV MTases are. Furthermore, we delineate the configuration of ZIKV MTase, including its domain hierarchy, amino acid compositions, binding sites, and post-translational modification (PTM). This work emphasizes the need to advance fundamental scientific understanding of virus families.
Recent ZIKV infection outbreaks in America have shown that this newly discovered arbovirus disease is a possible cause of congenital anomalies in children of infected mothers. The rapid spread of ZIKV, which has spread to Asian countries, including those bordering India, is a cause for concern at the global level. The available tools Prosite, Pfam, and InterProScan help locate the MTase domain. In addition to alignment, amino acid composition of, charged amino acids, atomic level studies & molecular weight, we correspondingly make predictions for these variables, including theoretical Pi. We also examine the modeled MTase domain structure (helices, beta turns) and its peculiarities, including secondary structure. We also discussed where proteins, DNA, and RNA bind. We also examined proteins, DNA & RNA bind. In a recent study, we evaluated the specifics of the binding sites, helices, beta turns, and structure (atomic and amino acid). In addition, we locate the protein, DNA, and RNA critical regions of the MTase domain, which may contain phosphorylation sites at Ser, Thr, & Tyr residues. These results suggest a complicated interaction between different phosphorylation modifications that modulate the activity of the MTase domain. An important goal of future research will be to fully understand the complementary and appropriate aspects and define the multiple roles that PTM plays in the MTase domain.
Acknowledgments
The authors express heartfelt gratitude towards the Department of Pharmaceutics, College of Pharmacy, Prince Sattam Bin Abdulaziz University, Alkharj 11942, Saudi Arabia.
Conflict of Interest
The authors declare that there is no conflict of interest.
BA, SA, & NHS: designed & first drafted the manuscript. AA, AMA, & AHA: data collection & editing of the manuscript; MYA and AAY: literature survey and revision; SSA, AMF: data collection, MAK designed experiments, final manuscript drafting, & reviewed the manuscript. The final draft was read and approved by all authors.
![]() References
References
- Yadav S, Rawal G, Baxi M. An overview of the latest infectious diseases around the world. The Journal of Community Health Management, (2016); 3(1):41-43.
- Yadav S, Rawal G. The current mental health status of Ebola survivors in Western Africa. Journal of Clinical and Diagnostic Research, (2015); 9(10): LA01-2.
- Yadav S, Rawal G, Baxi M. Zika virus -A pandemic in progress. Journal of Translational Internal Medicine, (2016); 4 (1):42-5.
- Chang C, Ortiz K, Ansari A, Gershwin ME. The Zika outbreak of the 21 st century. Journal of Autoimmunity, (2016); 68:1-13.
- Malone RW, Homan J, Callahan MV, Glasspool-Malone J, Damodaran L, Schneider A, et al. Zika Virus: Medical Countermeasure Development Challenges. PLOS Neglected Tropical Diseases, (2016); 10 (3): e0004530.
- Sikka V, Chattu VK, Popli RK, Galwankar SC, Kelkar D, Sawicki SG, et al. The Emergence of Zika Virus as a Global Health Security Threat: A Review and a Consensus Statement of the INDUSEM Joint working Group (JWG). Journal of Global Infectious Diseases, (2016); 8 (1): 3-15.
- Mehrjardi MZ Is Zika Virus an Emerging TORCH Agent? An Invited Commentary. Virology, (2017); 8:1178122X17708993.
- Lindenbach BD, Rice CM. Molecular biology of flaviviruses. Advances in Virus Research, (2003); 59, 23-61.
- Sirohi D, Kuhn RJ. Zika Virus Structure, Maturation, and Receptors. Journal of infectious diseases, (2017); 216, (suppl_10): S935-S944.
- Issur M, Geiss BJ, Bougie I, Picard-Jean F, Despins S, Mayette J, Hobdey SE, Bisaillon M. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA, (2009); 15 (12): 2340-2350.
- Piorkowski G, Richard P, Baronti C, Gallian P, Charrel R, Leparc-Goffart I, Lamballerie XD. Complete coding sequence of Zika virus from Martinique outbreak in 2015. New Microbes and New Infections, (2016); 11, 52-53.
- Elshahawi H, Syed Hassan S, Balasubramaniam V. Importance of Zika Virus NS5 Protein for Viral Replication. Pathogens, (2019); 8 (4): 169.
- Godoy AS, Lima G M, Oliveira KI, Torres NU, Maluf FV, Guido RV, Oliva G. Crystal structure of Zika virus NS5 RNA-dependent RNA polymerase. Nature Communications, (2017); 8, 14764.
- Zhang C, Feng T, Cheng J, Li Y, Yin X, Zeng W, Jin X, Li Y, Guo F, Jin T. Structure of the NS5 methyltransferase from Zika virus and implications in inhibitor design. Biochemical and Biophysical Research Communications, (2017); 492 (4): 624-630.
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server; (In) John M. Walker (ed): The Proteomics Protocols Handbook, Humana Press (2005). 571-607.
- Boeckmann B, Bairoch A, Apweiler R, Blatter M-C, Estreicher, A, Gasteiger E, Martin MJ, Michoud K, O’Donovan C, Phan I, Pilbout S, Schneider M. The Swiss-Prot protein knowledgebase and its supplement TrEMBL in 2003. Nucleic acids research, (2003); 31(1): 354-370.
- Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger, E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh L-SL. UniProt: the Universal Protein knowledgebase. Nucleic acids research, (2004); 432, D115-D119.
- Wu S, Skolnick J, Zhang Y Ab initio modeling of small proteins by iterative TASSER simulations. BMC biology, (2007); 5: 17.
- Roy A, Kucukural A, Zhang Y I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols, (2010); 5 (4): 725-738.
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. Journal of molecular biology, (1999); 292 (2): 195-202.
- Wu S, Zhang Y LOMETS: a local meta-threading-server for protein structure prediction. Nucleic acids research, (2007); 35 (10): 3375-3382.
- Zhang Y, Kihara D, Skolnick J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins: Structure, Function, and Bioinformatics, (2002); 48 (2): 192-201.
- Wu S, Zhang Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics, (2008); 24 (7): 924-931.
- Zhang Y, Skolnick J. SPICKER: A clustering approach to identify near‐native protein folds. Journal of computational chemistry, (2004); 25 (6): 865-871.
- Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic acids research, (2005); 33 (7): 2302-2309.
- Li Y, Zhang Y. REMO: A new protocol to refine full atomic protein models from C‐alpha traces by optimizing hydrogen‐bonding networks. Proteins: Structure, Function, and Bioinformatics, (2009); 76 (3): 665-676.
- Lovell SC, Davis IW, Arendall WB, de Bakker PI, Word JM, et al. (2003) Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins: Structure, Function, and Bioinformatics, (2003); 50 (3): 437-450.
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. Journal of molecular biology, (1999); 292(2): p. 195-202.
- Geourjon C, Deléage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comparative Study. Computer Applications in the Biosciences, (1995); 11(6):681-4.
- Yachdav G, Kloppmann E, Kajan L, Hecht M, Goldberg T, Hamp T, Hönigschmid P, Schafferhans A, Roos M, Bernhofer M, Richter L, Ashkenazy H, Punta M, Schlessinger A, Bromberg Y, Schneider R, Vriend G. PredictProtein – Predicting Protein Structure and Function for 29 Years. Nucleic Acids Research, (2021);49(W1):W535-W540
- Vittal R.Srinivas, Bioinformatics-A Practical Guide to the analysis of Genes and Proteins” , (2005); ISBN : 978-81-203-2858-7.
- Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S: Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics, (2004); 4(6):1633-1649.
- Minguez P, Letunic I, Parca L, Bork P: PTMcode: a database of known and predicted functional associations between post-translational modifications in proteins. Nucleic acids research, (2012); 41(D1): D306-D311.
- Shahin Ramazi and Javad Zahiri. Post-translational modifications in proteins: resources, tools and prediction methods. Database (Oxford), (2021); baab012.
- Ramazi S, Allahverdi A, and Zahiri J. Evaluation of post-translational modifications in histone proteins: a review on histone modification defects in developmental and neurological disorders. Journal of Biosciences, (2020); 45, 135.
- Mann M, and Jensen ON. Proteomic analysis of post-translational modifications. Nature Biotechnology, (2003); 21(3): 255-261.
- Chen LH & Hamer DH. Zika Virus: Rapid Spread in the Western Hemisphere. Annals of Internal Medicine, (2016); 164(9):613-5.
- Triunfol M. A new mosquito-borne threat to pregnant women in Brazil, The Lancet Infectious Diseases, (2016); 16(2):156-7.
- Lim SP, Noble CG and Shi P-Y. The dengue virus NS5 protein as a target for drug discovery. Antiviral Research, (2015); 119: 5767.
- Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nature Protocol, (2008); 3:1171-1179.
- Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, Womack TO. BUSTER version 2.11.2. Global Phasing Ltd, Cambridge, United Kingdom, (2011).
- Lim SP, Sonntag LS, Noble C, Nilar SH, Ng RH, Zou G, Monaghan P, Chung KY, Dong H, Liu B, Bodenreider C, Lee G, Ding M, Chan WL, Wang G, Jian YL, Chao AT, Lescar J, Yin Z, Vedananda TR, Keller TH, Shi PY. Small molecule inhibitors that selectively block dengue virus methyltransferase. Journal of Biological Chemistry, (2011); 286 (8):6233-6240.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0