

Review Article
Current advances on single or multi-omics analysis of esophageal cancer
Kaidirina Kasimu1, Wenwen Cui2, Yihan Wang2, Xin Li3, Hongbo Wang4,5,6*, Xiaotong Yu4,5,6, Fu Ren4,5,6*
Adv. life sci., vol. 11, no. 2, pp. 296-304, May 2024
*– Corresponding Authors: Hongbo Wang (18640249580@163.com)
Fu Ren (rf@symc.edu.cn)
Authors' Affiliations
2. School of basic medicine, Shenyang Medical College, Shenyang – P.R. China
3. School of Stomatology, Shenyang Medical College, Shenyang – P.R. China
4. Department of Human Anatomy, School of basic medicine, Shenyang Medical College, Shenyang – P.R. China
5. Key laboratory of Human Ethnic Specificity and Phenomics of Critical Illness in Liaoning Province – P.R. China
6. Key Laboratory of Phenomics Research in Shenyang – P.R. China
[Date Received: 01/12/2024; Date Revised: 15/02/2024; Date Available Online: 18/04/2024]
Editorial Note: You are viewing the latest version of this manuscript having minor correction in roles of authors, which are different from originally published copy.
Abstract![]()
Introduction
Methods
Discussion
Conclusion
References
Abstract
Esophageal cancer is associated with high mortality rates and is one of the cancers with the worst prognosis. Its incidence has significant regional specificity, particularly in China where it is much higher than in other countries. Moreover, effective diagnostic markers, therapeutic targets, and molecular subtyping biomarkers are currently lacking for esophageal cancer. Nevertheless, large-scale omics studies have identified dozens of robust genetic risk loci and prognosis-related loci, drawn genomic, epigenomic, and transcriptomic maps of esophageal cancer at multiple molecular levels, and described significant differences between esophageal squamous cell carcinoma and adenocarcinoma. These studies are of great significance for exploring the occurrence and development mechanism of esophageal cancer, guiding clinical treatment, and improving patient prognosis. This review, from the perspective of multi-omics, discusses the analytical strategies employed in these studies and summarizes their core findings. It emphasizes that the integration and analysis of multi-omics data is a key focus and development trend in the precise medical research of esophageal cancer, and has broad research and application prospects.
Keywords: Esophageal cancer; GWAS; Precision medicine; Biomarker
Introduction![]()
Esophageal cancer has a high incidence and mortality rate, and exhibits significant regional variation. Although the incidence of esophageal cancer in China has been declining, it remains significantly higher than in other countries [1–4]. Currently, effective therapeutic targets for esophageal cancer are lacking, making it one of the deadliest cancers in China [1]. Esophageal cancer is primarily categorized as squamous cell carcinoma (ESCC) or adenocarcinoma (EAC), with squamous cell carcinoma being more prevalent. Squamous cell carcinoma of the esophagus is characterized by invasiveness and has a significantly worse prognosis than adenocarcinoma. These two cancers differ significantly in histopathology, incidence, patient population, and prognosis, leading some to suggest that they should be regarded as two distinct diseases [5]. The pathogenesis of esophageal cancer remains incompletely understood, and there is a dearth of therapeutic targets and biomarkers with clinical applicability.
In recent years, significant advancements have been made in second-generation sequencing and gene chip technology. Correspondingly, precision medicine research has become a hot area of investigation that utilizes omics analysis techniques to aid in the more refined treatment of complex human diseases such as cancer. This article reviews various omics analysis studies conducted on esophageal cancer in recent years. These studies have identified many molecular targets highly associated with esophageal cancer at various molecular levels. These results are significant in identifying susceptible loci and carcinogenic genes for esophageal cancer, providing potential targets for clinical treatment, and improving patient prognosis. However, there is still a lack of comprehensive research that integrates multiple omics results to systematically explain the pathogenesis of esophageal cancer. In conclusion, second-generation sequencing and gene chip technology have broad prospects for the research and clinical treatment of esophageal cancer.
Methods![]()
Literature Search and Selection Criteria
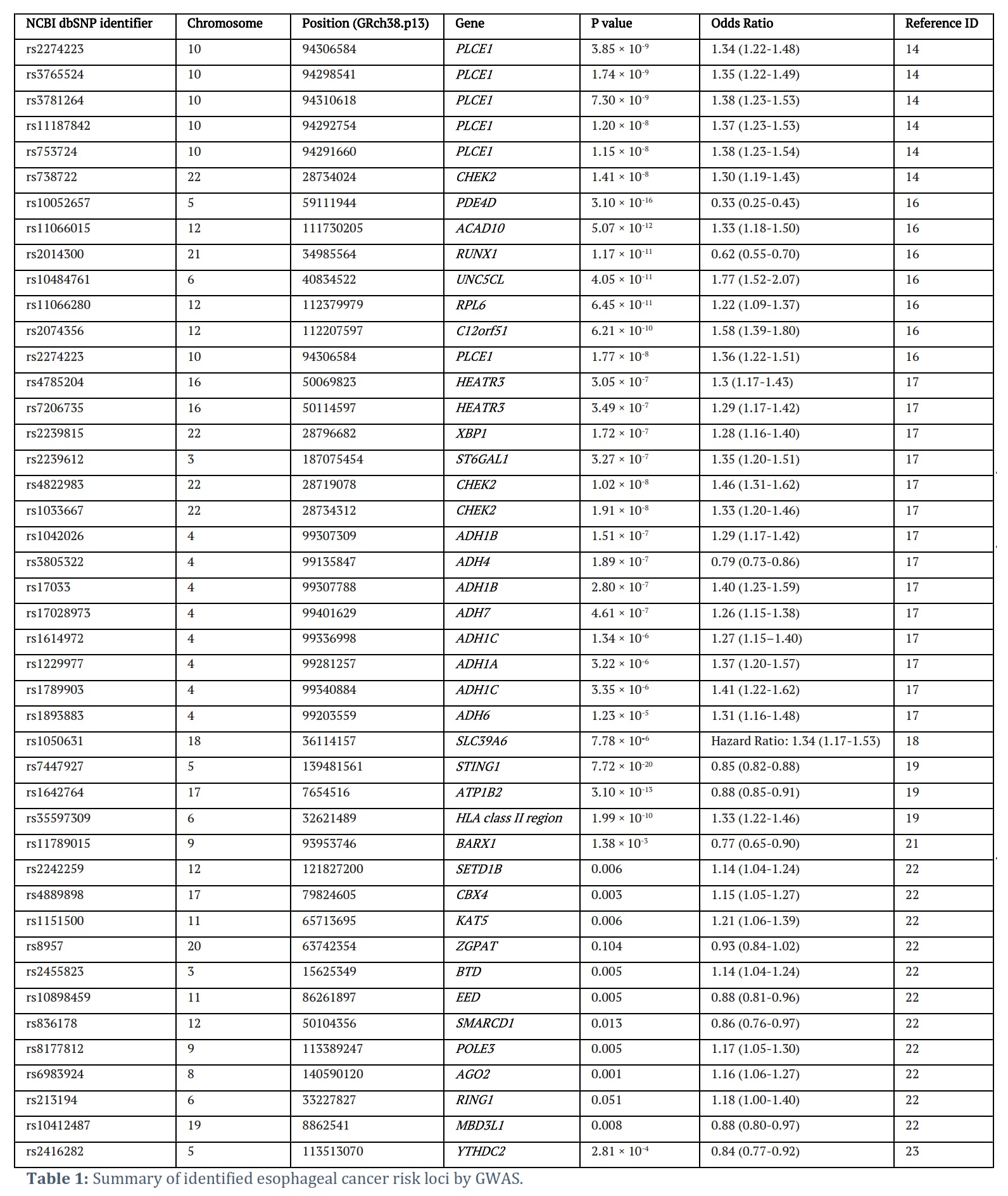
This article summarizes recent studies on omics analysis related to esophageal cancer. It highlights the identification of numerous molecular targets strongly linked to the disease across different molecular layers, including rs1050631, which shows a strong correlation with patient outcomes, and mutations in genes like TP53, CDKN2A, PIK3CA, detected in esophageal cancer samples (Table 1). These findings are crucial for pinpointing risk loci and oncogenes in esophageal cancer, offering possible avenues for therapy and enhancing the prognosis for patients.
Discussion![]()
Genetic Risk Research of Esophageal Cancer Based on GWAS
Esophageal cancer is a complex, multi-gene phenotype that can be heritable, with a heritability estimate ranging from 19% to 35% [6–12]. GWAS is an effective method for discovering specific genetic risk loci. These studies typically use the whole-genome genotyping data of a large cohort of esophageal cancer patients and healthy controls to test the correlation of numerous genetic loci with the risk or prognosis of the disease. This enables the identification of candidate loci and genes.
Currently, there are many reports on large-scale GWAS studies of esophageal cancer in the Chinese population, which can be explained by the high incidence of esophageal cancer in this population [13,14]. To date, dozens of robust ESCC-associated genetic regions and candidate genes have been discovered, such as rs2274223, rs3765524, rs3781264, and rs11187842 (Table 1). The earliest GWAS study collected 2,115 ESCC patients and 3,302 healthy Chinese controls and identified multiple genetic susceptibility loci on PLCE1 located on chromosome 10q23 [15]. This finding was further validated by another independent study [16]. Subsequently, seven risk loci for ESCC were identified on chromosomes 5q11, 6p21, 10q23, 12q24, and 21q22 [17].
In a GWAS study including 2,031 ESCC patients and 2,044 healthy Chinese controls, Wu et al. identified six new ESCC susceptibility loci, four of which were associated with ESCC risk, and the other two loci were only associated with ESCC risk when considering gene-alcohol interactions [18]. Later, Wu et al. found that rs1050631 on SLC39A6 was significantly associated with the survival of ESCC patients [19]. Wu et al. then attempted to merge and analyze several large ESCC cohorts and discovered two new ESCC risk loci, rs7447927 and rs1642764, which were not found in previous independent cohorts [15–17, 20].
Finally, Wang et al. compiled a list of the 24 most significant Chinese ESCC risk SNPs identified in previous studies and further validated them [21]. Interestingly, Yan et al. found that the genetic variant rs11789015 on 9q22 may be associated with the risk of both EAC/BE and ESCC, and this correlation may be achieved by regulating the function of BARX1, indicating that there may be a certain degree of shared genetic risk loci between EAC and ESCC [22].
Post-GWAS analysis and interpretation were mainly conducted based on epigenetic modification-associated loci identified by GWAS analysis.
Sung et al. utilized GWAS to identify genetic loci on epigenetic modification-associated genes (particularly chromatin remodeling) that were associated with ESCC risk [23]. In addition, Yang et al. found that rs2416282 led to ESCC risk by regulating the expression of YTHDC2, which controls RNA m6A modification. This study provided us with a new idea that genetic risk loci may affect the occurrence and development of ESCC through RNA m6A modification [24].
Several studies have demonstrated that certain methylation modifications can be inherited. However, most EWAS studies have found that changes in methylation modifications are mainly due to environmental factors, such as age and smoking. It is currently unclear whether specific methylation sites can influence the genetic risk of esophageal cancer. This significant scientific question deserves further exploration through methods such as EWAS analysis. In summary, large-scale GWAS analysis of esophageal cancer cohorts has made progress in identifying many esophageal cancer risk loci, while EWAS research is a direction worthy of continued attention in the future.
Several studies have demonstrated that certain methylation modifications can be inherited. However, the majority of EWAS studies have found that changes in methylation modifications are primarily the result of environmental factors, such as age and smoking. Currently, it remains unknown whether specific methylation sites can influence the genetic risk of esophageal cancer. This critical scientific question necessitates further exploration through methods such as EWAS analysis. In conclusion, large-scale GWAS analysis of esophageal cancer cohorts has made considerable progress in identifying many esophageal cancer risk loci, while EWAS research is a direction that deserves continued attention in the future.
Post-GWAS functional annotation of esophageal cancer based on eQTL analysis:
Multi-omics techniques are mainly focused on the in-depth interpretation of GWAS results, which is known as post-GWAS analysis. Identification and analysis of expression quantitative trait loci (eQTLs) is a typical representative of post-GWAS analysis. Expression quantitative traits refer to the continuous variation in the expression of a gene in the entire population, which is controlled by multiple genetic loci and is easily influenced by environmental factors. eQTLs refer to the set of SNP loci that control gene expression. Similar loci include methylation QTL (meQTL) and protein QTL (pQTL), based on which genome-wide SNP-gene, SNP-CpG, and SNP-protein interaction maps can be constructed. Currently, eQTL research is the most extensive, and eQTL maps of various healthy tissues have been constructed. However, research on meQTL and pQTL has only recently begun to emerge.
To perform post-GWAS analysis, which uses eQTL as a representative, researchers typically establish large cohorts of both esophageal cancer patients and healthy individuals, collect peripheral blood samples, and identify germline mutation sites associated with the phenotype.
They then often conduct gene expression and other multi-omics level analyses and identifications in public databases or a small number of tissue samples. Ultimately, they obtain eQTL sites that are relevant to the occurrence and development of esophageal cancer, thereby explaining the functional role of the SNPs identified in GWAS analysis or identifying eQTL sites associated with the risk or prognosis of esophageal cancer.
Currently, researchers investigating esophageal cancer typically identify eQTL sites on genes associated with esophageal squamous cell carcinoma or adenocarcinoma (such as CUL3, CFDP1, SLC22A3) using SNPs identified in GWAS analysis. These eQTL sites are then validated in vitro experiments or normal esophageal tissues, or through the use of public databases such as GTEx [23,25,26]. Additionally, Shao et al. identified SNPs related to the miR-15 family and found that the miR-15b SNP rs1451761T>G was significantly associated with a reduced risk of ESCC, with this association being influenced by smoking
status [27]. Cui et al. used GWAS and eQTL analysis to identify a gene variant, rs1154402C>G, that inhibits alcohol dehydrogenase gene ADH1A expression, leading to susceptibility to esophageal squamous cell carcinoma [28].
Both studies once again confirm the impact of smoking and alcohol consumption on the occurrence and development of esophageal cancer, which is consistent with previous research findings.
It is worth noting that Peng et al. have developed the Chinese Cancer Genome Database for Esophageal Squamous Cell Carcinoma (CCGD-ESCC) (http://db.cbi.pku.edu.cn/ccgd/ESCCdb). The database contains 69,593 SNPs, including risk SNPs identified from 2,022 cases of cancer and 2,039 controls, survival-related SNPs identified from 1,006 cases of esophageal squamous cell carcinoma (survival GWAS), and eQTLs identified from the expression profiles of 94 cases of esophageal squamous cell carcinoma. Additionally, the database provides information on the relationship between 8,833 somatic mutations and survival time in 675 patients with esophageal squamous cell carcinoma [29].
To summarize, multi-omics studies on large-scale cohorts of esophageal cancer represent an emerging field. Currently, the primary focus is on the combined analysis of GWAS and eQTL data. These studies have uncovered new loci from a novel perspective while remaining consistent with the primary findings of conventional GWAS analyses. However, there are no reported post-GWAS functional annotations of other omics data. Therefore, further exploration in this area is necessary, including meQTL and pQTL, as well as SNP-CpG and SNP-protein interaction maps of large-scale cohorts of esophageal cancer.
Single-Omics-Based Studies of Cancer and Adjacent Tissues:
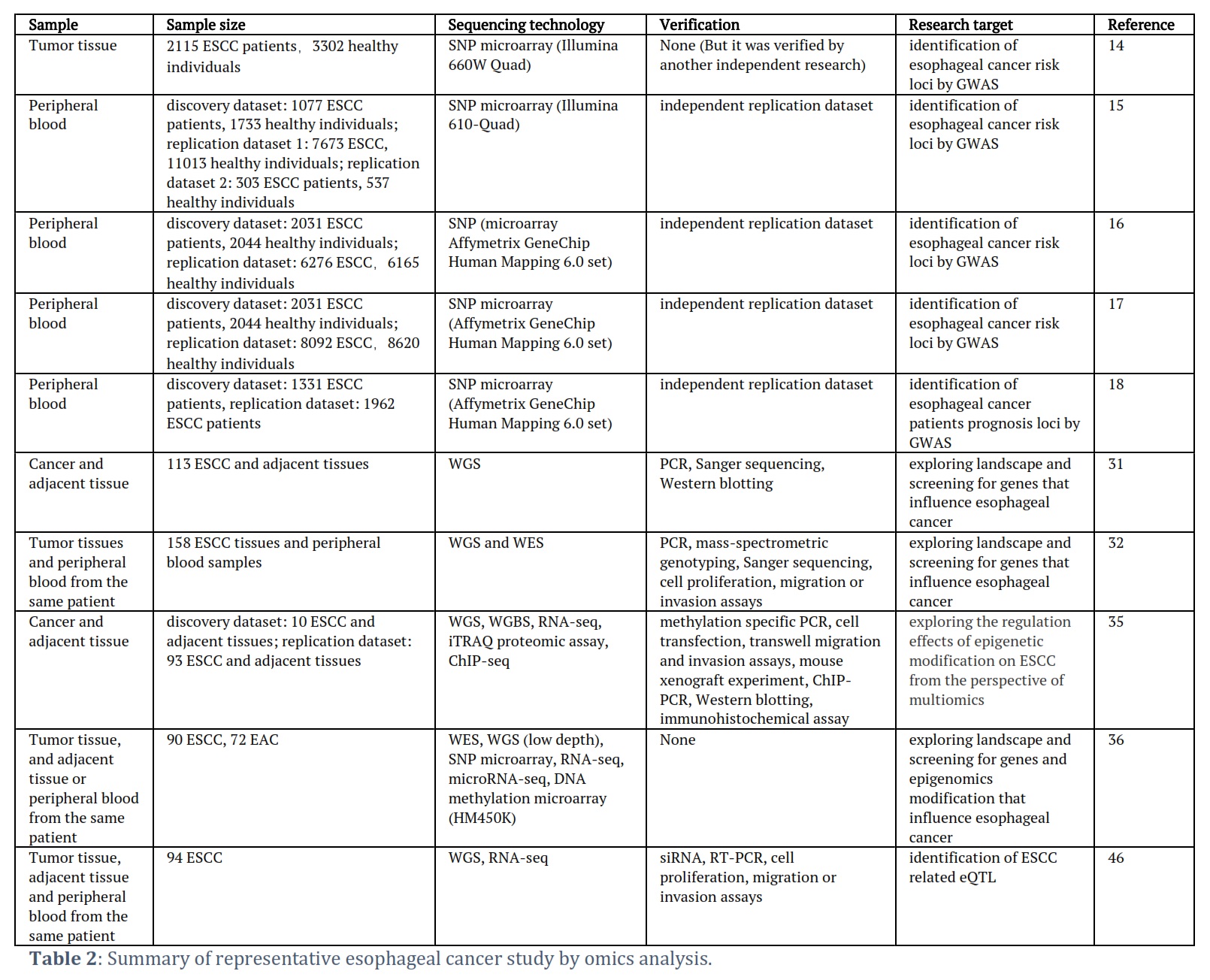
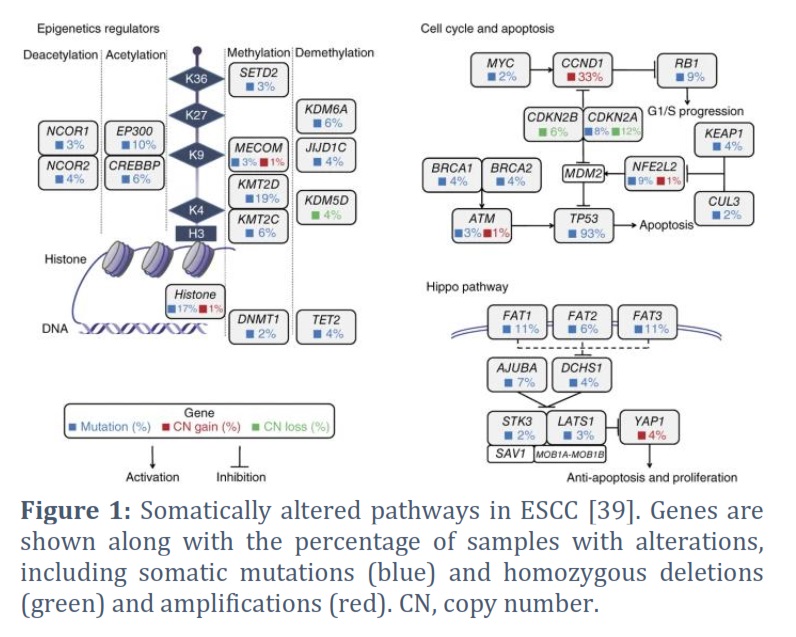
In addition to genetic mutations, somatic mutations unique to cancer tissue often receive significant attention compared to healthy tissue. From a non-genetic perspective, a typical experimental approach involves collecting a specific number of cancer and adjacent tissue samples and studying the differential omics profiles of esophageal cancer and adjacent tissue. This approach aims to identify driver genes, abnormally expressed genes in cancer tissue, and aberrantly regulated pathways. Ultimately, mutated or differentially expressed genes related to tumor occurrence and development, molecular subtypes, or patient prognosis are identified and considered potential therapeutic targets or tumor biomarkers. In 2012, Agrawal et al. conducted the first whole-genome mutation study of esophageal cancer by comparing esophageal squamous cell carcinoma (ESCC) and adenocarcinoma. The study showed that NOTCH1 mutations were unique to ESCC and were not found in EAC. Furthermore, most EAC mutations could also be found in the paired normal Barrett's esophagus, which has the potential to develop adenocarcinoma [30]. In 2014, Gao et al. published their study on the exome landscape of ESCC, revealing numerous somatic mutations involving genes that regulate cell cycle and apoptosis (TP53, CCND1, CDKN2A, NFE2L2, RB1), as well as multiple mutations in genes associated with histone modification (KMT2D, KMT2C, KDM6A, EP300, CREBBP) (Figure 1). Additionally, the study found that mutations in FAT1, FAT2, FAT3, FAT4, and AJUBA led to abnormal regulation of the Hippo pathway, while mutations in NOTCH1, NOTCH2, NOTCH3, and FBXW7 resulted in aberrant regulation of the Notch pathway in ESCC [31]. In the same year, Yongmei Song et al. identified eight significantly mutated genes in ESCC through whole-genome and whole-exome sequencing, including six well-known tumor-related genes (TP53, RB1, CDKN2A, PIK3CA, NOTCH1, NFE2L2) and two tumor-related genes specific to ESCC (ADAM29 and FAM135B). Functional experiments confirmed that FAM135B promoted ESCC deterioration, while MIR548K was identified to promote the same. Mutations in several genes associated with histone modification (KMT2D, ASH1L, KMT2C, SETD1B, CREBBP, and EP300) were also found, along with abnormalities in the Wnt, cell cycle, and Notch pathways. The study also established a significant correlation between alcohol consumption and the development of ESCC [32]. In 2016, Sawada et al. conducted a study on a Japanese population of patients with ESCC and arrived at similar conclusions [33]. In 2020, Cui et al. determined the whole-genome of 508 ESCC tumor tissues, identifying five novel esophageal cancer-related mutation genes (KRT5, CDH10, LILRB3, YEATS2, and CASP8). Additionally, they found that NFE2L2 may be a tumor suppressor gene for ESCC, and its mutations were significantly associated with the poorest prognosis for ESCC. Furthermore, since the range of detection for whole-genome sequencing is much greater than that of previous exome sequencing, this study also found that non-coding mutations in the SLC35E2 gene promoter region were significantly associated with poor prognosis in ESCC [34]. The aforementioned studies have mapped the genomic profiles of esophageal squamous cell carcinoma and adenocarcinoma, identifying subtype-specific gene mutations and abnormal signaling pathways. These results provide the groundwork for subsequent omics analysis of esophageal cancer and targeted therapy in clinical settings.
Integrated Omics Analysis for Differential Studies of Cancer and Adjacent Tissues:
At the single omics level, differential genes often fail to exhibit differences at other omics levels. However, with advancements in sequencing technologies and cost reduction, integrated omics analysis has become more prevalent. By utilizing multi-omics techniques to generate multi-omics differential profiles of cancer and adjacent tissues and conducting integrated analysis, more reliable multi-omics differential genes can be obtained that are related to tumor occurrence and development, molecular subtypes, or patient prognosis compared to single-omics results. These genes have a stronger functional interpretation, and their mechanisms of action are better understood, making them more reliable potential therapeutic targets or tumor markers.
Multi-omics analysis of esophageal cancer is currently focused on integrating differentially expressed genes, DNA methylation, and histone modification data to explore the epigenetic regulatory mechanisms underlying the occurrence and development of esophageal cancer [35]. Typical abnormal genes identified include SOX2, CCND1, TP53, PIK3CA, and NOTCH1, while common abnormal regulatory pathways include the Hedgehog signaling and PI3K pathways. In 2017, the TCGA research team conducted a comprehensive analysis of 164 esophageal cancer samples from Eastern and Western populations, which included copy number variations, mRNA, microRNA, and DNA methylation. The four omics were subjected to unsupervised learning clustering, and the clustering results were consistent and matched the histopathological classification of esophageal squamous cell carcinoma and adenocarcinoma. In esophageal squamous cell carcinoma, the genes CCND1, SOX2, and/or TP63 are frequently amplified, while ERBB2, VEGFA, GATA4, and GATA6 are more commonly amplified in adenocarcinoma. Compared to esophageal adenocarcinoma, the molecular patterns of esophageal squamous cell carcinoma are more similar to those of squamous cell carcinoma in other organs. These results suggest that different treatment approaches should be adopted clinically for esophageal squamous cell carcinoma and adenocarcinoma [36].
Ping et al. has conducted a series of studies to identified the large number of somatic structural variations (SV) and gene mutations (APOBEC, PIK3CA, ERBB4, BRCA1/2, etc.) in esophageal squamous cell carcinoma [37–39]. In around 60% of cases, hedgehog signaling and the PI3K pathway are highly active, suggesting that targeting these pathways could be a promising strategy for treating ESCC [37]. Furthermore, in a small number of patients, amplification of CD274 leads to high expression of PD-L1, indicating the potential for immune therapy in these patients [38]. Using a multi-region whole-exome sequencing approach, Chen et al. conducted a study on the progression from esophageal squamous epithelial hyperplasia precursor lesions to ESCC and found that complete inactivation of TP53 plays a significant promoting role in ESCC development [40]. Similarly, Lin et al. identified a large number of somatic copy number variations in ESCC, including FAT1, FAT2, ZNF750, KMT2D, and previously identified TP53, PIK3CA, and NOTCH1, and found that multiple molecular mechanisms regulating the PI3K pathway are disrupted in ESCC. They also observed gene mutation and protein-level overexpression of XPO1 in ESCC, indicating that XPO1 has high potential for targeted therapy [41]. In addition, they compared the molecular characteristics of esophageal squamous cell carcinoma and adenocarcinoma in detail using exome sequencing, whole-genome methylation sequencing, and ChIP-seq, and identified two nearly independent driver gene sets in ESCC and EAC, respectively. This suggests that they follow independent developmental pathways, consistent with the results of previous studies. Moreover, the study identified two ESCC-specific tumor suppressor genes, CUL3 and ZFP36L2 [42].
Qin et al. conducted a study using whole-genome and whole-exome sequencing and identified mutations in the VANGL1 gene. Furthermore, they found that this gene can promote cell growth in vitro. Additionally, the study revealed five genes with somatic copy number alterations (SCNA) or structural variations (SV), including three coding genes (SHANK2, MYBL2, FADD) and two non-coding genes (miR-4707-5p, PCAT1). Based on the expression profiles of 321 ESCC individuals, survival analysis showed a significant correlation between these genes and a poor survival rate. Subsequent functional experiments validated the results of the bioinformatics analysis and demonstrated that miR-4707-5p and MYBL2 promote tumor proliferation and metastasis [43]. The studies mentioned above (Table 2) provide a systematic exploration of the mechanisms underlying the occurrence and development of esophageal cancer by integrating data from multiple omics levels. They identify several genes that are significantly associated with patient prognosis, which can serve as candidate genes for targeted therapy.
Exploring Factors that Influence the Occurrence and Development of Esophageal Cancer:
Studies on mutation clones in normal esophageal epithelium at different ages have identified NOTCH1 and TP53 mutations that accumulate with increasing age. Moreover, smoking and alcohol consumption can significantly accelerate this accumulation, indicating that lifestyle may play a role in the onset and progression of esophageal cancer [44,45]. In addition, Chang et al.’s whole-genome sequencing and transcriptome sequencing studies have revealed that ESCC is linked to genetic variations in alcohol intake and metabolism enzymes. They also identified abnormal cell cycle and PI3K-Akt pathway in ESCC, which are consistent with previous research findings [46]. The incidence of esophageal adenocarcinoma (EAC) and its precursor lesion, Barrett’s esophagus (BE), is significantly higher in men than in women. Dong et al. screened over 9 million genetic variations and found that rs112894788 was significantly associated with the risk of BE/EAC only in male individuals, while rs13259457 was only significantly associated with the risk of BE/EAC in female individuals [47]. Esophageal cancer is known for its high incidence rates and specific racial and regional patterns, highlighting the need for a thorough comparison of incidence rates and survival patterns between different races. Deng et al. conducted whole-exome and targeted sequencing on samples from 316 Chinese patients with esophageal squamous cell carcinoma and compared their findings to data from European patients in TCGA. The study revealed that Asian patients with CSMD3 mutations had a better prognosis, and TP53, EP300, and NFE2L2 had higher mutation frequencies in Asian patients. This research sheds light on the molecular mechanisms that underlie the racial differences in esophageal cancer incidence rates [48].
Hao et al.'s study on spatial heterogeneity and clonal evolution in ESCC revealed that around 35.8% of somatic mutation heterogeneity in ESCC originated from spatial heterogeneity. Half of the driver mutations on the branches of the esophageal cancer phylogenetic tree targeted oncogenes such as PIK3CA, NFE2L2, and MTOR, among others. In contrast, the majority of truncal and clonal driver mutations were observed in tumor suppressor genes such as TP53, KMT2D, and ZNF750, among others [49].
The studies mentioned above demonstrate that various factors such as age, smoking, alcohol consumption, racial specificity, and spatial heterogeneity can impact the sequencing data results of the esophageal cancer genome. Therefore, when conducting in-depth large-scale esophageal cancer omics data analyses in the future, it is crucial to consider including these factors as covariates to ensure the accuracy of subsequent data analysis results.
Figures & Tables
Esophageal cancer is characterized by a high incidence and mortality rate, and limited treatment options, which are even more evident in China. It is influenced by various environmental factors such as age, smoking, and alcohol consumption, and exhibits racial specificity. The subtypes of esophageal cancer have distinct molecular patterns, necessitating personalized targeted therapies for each subtype. Current second-generation sequencing studies, based on single-omics or multi-omics data analysis, have identified numerous unique genetic or non-genetic variant genes and abnormal pathways specific to esophageal cancer, providing a crucial foundation for subsequent treatment and improved patient prognosis. However, post-GWAS multi-omics analyses of esophageal cancer, especially in QTL research, are limited to the eQTL field, and other multi-omics integrated research based on large-scale individual data is relatively rare. Such research can demonstrate multidimensional molecular level interactions in the whole genome, thereby building causal regulatory networks from genes, epigenetic modifications to expression, and translation of proteins, and showing the association of multidimensional sites with the incidence and prognosis of esophageal cancer. In summary, cohort-based multi-omics analysis is the future trend of precision medicine research on esophageal cancer and is worthy of further exploration.
Conflict of Interest
The authors declare no conflict of interest.
Funding Information
This study was support by Center Guiding Local Science and Technology Foundation of Liaoning Science and Technology Committee (2023JH6/100100021) to Fu Ren, the Technology Department of Liaoning Province (No. 2023JH2/101300079) to Xin Li, the Education Department of Liaoning Province (JYTMS20231395) to Hongbo Wang, and College Students’ Innovation and Training Program of Shenyang Medical College (X202310164020) to Wenwen Cui.
K.K. and W.C.: investigation and collection of data. K.K., X.L. and H.W.: analysis and original draft. K.K. and X.Y.: reviewing and editing the manuscript. F.R.: conceptualization, supervision.: K.K., reviewing and editing the manuscript.
![]() References
References
- Chen W, Zheng R, Baade PD, Zhang S, Zeng H. et al. Cancer statistics in China, 2015: Cancer Statistics in China, 2015. CA: A Cancer Journal for Clinicians (2016); 66:115–132.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA A Cancer Journal for Clinicians, (2019); 69:7–34.
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J. et al. Global cancer statistics, 2012: Global Cancer Statistics, 2012. CA: A Cancer Journal for Clinicians (2015); 65:87–108.
- Li H, Mu F, Zou B, Wang L. Pulmonary sarcoidosis-like reactions induced by sintilimab in esophageal cancer: A case report. Medicine, (2023); 102: e34432.
- Siewert JR, Ott K. Are Squamous and Adenocarcinomas of the Esophagus the Same Disease? Seminars in Radiation Oncology, (2007); 17:38–44.
- Dai J, Shen W, Wen W, Chang J, Wang T. et al. Estimation of heritability for nine common cancers using data from genome-wide association studies in Chinese population: Estimation of heritability for nine common cancers. International Journal of Cancer, (2017); 140:329–336.
- Contino G, Vaughan TL, Whiteman D, Fitzgerald RC. The Evolving Genomic Landscape of Barrett’s Esophagus and Esophageal Adenocarcinoma. Gastroenterology, (2017); 153:657-673.e1.
- Ek WE, Levine DM, D’Amato M, Pedersen NL, Magnusson PKE. et al. Germline Genetic Contributions to Risk for Esophageal Adenocarcinoma, Barrett’s Esophagus, and Gastroesophageal Reflux. JNCI Journal of the National Cancer Institute, (2013); 105:1711–1718.
- Gharahkhani P, Fitzgerald RC, Vaughan TL, Palles C, Gockel I. et al. Genome-wide association studies in oesophageal adenocarcinoma and Barrett’s oesophagus: a large-scale meta-analysis. The Lancet Oncology, (2016); 17:1363–1373.
- Levine DM, Ek WE, Zhang R, Liu X, Onstad L. et al. A genome-wide association study identifies new susceptibility loci for esophageal adenocarcinoma and Barrett’s esophagus. Nature Genetics, (2013); 45:1487–1493.
- Palles C, Chegwidden L, Li X, Findlay JM, Farnham G. et al. Polymorphisms Near TBX5 and GDF7 Are Associated With Increased Risk for Barrett’s Esophagus. Gastroenterology, (2015); 148:367–378.
- The Esophageal Adenocarcinoma Genetics Consortium, The Wellcome Trust Case Control Consortium 2, Su Z, Gay LJ, Strange A. et al. Common variants at the MHC locus and at chromosome 16q24.1 predispose to Barrett’s esophagus. Nature Genetics, (2012); 44:1131–1136.
- Hyland PL, Zhang H, Yang Q, Yang HH, Hu N. et al. Pathway, in silico and tissue-specific expression quantitative analyses of oesophageal squamous cell carcinoma genome-wide association studies data. Internatonal Journal of Epidemiology, (2016); 45:206–220.
- Tian J, Liu C, Liu G, Zuo C, Chen H. Cumulative evidence for association between genetic polymorphisms and esophageal cancer susceptibility: A review with evidence from meta‐analysis and genome‐wide association studies. Cancer Medicine, (2019); 8:1289–1305.
- Abnet CC, Freedman ND, Hu N, Wang Z, Yu K. et al. A shared susceptibility locus in PLCE1 at 10q23 for gastric adenocarcinoma and esophageal squamous cell carcinoma. Nature Genetics, (2010); 42:764–767.
- Wang L-D, Zhou F-Y, Li X-M, Sun L-D, Song X. et al. Genome-wide association study of esophageal squamous cell carcinoma in Chinese subjects identifies a susceptibility locus at PLCE1. Nature Genetics, (2010); 42:759–763.
- Wu C, Hu Z, He Z, Jia W, Wang F. et al. Genome-wide association study identifies three new susceptibility loci for esophageal squamous-cell carcinoma in Chinese populations. Nature Genetics, (2011); 43:679–684.
- Wu C, Kraft P, Zhai K, Chang J, Wang Z. et al. Genome-wide association analyses of esophageal squamous cell carcinoma in Chinese identify multiple susceptibility loci and gene-environment interactions. Nature Genetics, (2012); 44:1090–1097.
- Wu C, Li D, Jia W, Hu Z, Zhou Y. et al. Genome-wide association study identifies common variants in SLC39A6 associated with length of survival in esophageal squamous-cell carcinoma. Nature Genetics, (2013); 45:632–638.
- Wu C, Wang Z, Song X, Feng X-S, Abnet CC. et al. Joint analysis of three genome-wide association studies of esophageal squamous cell carcinoma in Chinese populations. Nat Genet (2014); 46:1001–1006.
- Wang K-L, Chen X-L, Lei L, Li P, Hong L-L. et al., Huang X-C, Mao W-M, Mukaisho K, Ling Z-Q. Validation study of susceptibility loci for esophageal squamous cell carcinoma identified by GWAS in a Han Chinese subgroup from Eastern China. Journal of Cancer, (2019); 10:3624–3631.
- Yan C, Ji Y, Huang T, Yu F, Gao Y. et al. An esophageal adenocarcinoma susceptibility locus at 9q22 also confers risk to esophageal squamous cell carcinoma by regulating the function of BARX1. Cancer Letters, (2018); 421:103–111.
- Sung H, Yang HH, Zhang H, Yang Q, Hu N. et al. Common genetic variants in epigenetic machinery genes and risk of upper gastrointestinal cancers. International Journal of Epidemiology, (2015); 44:1341–1352.
- Yang N, Ying P, Tian J, Wang X, Mei S Genetic variants in m6A modification genes are associated with esophageal squamous-cell carcinoma in the Chinese population. Carcinogenesis, (2020); 41:761–768.
- Hu JL, Hu XL, Lu CX, Chen XJ, Fu L Variants in the 3’-untranslated region of CUL3 is associated with risk of esophageal squamous cell carcinoma. Journal of Cancer (2018); 9:3647–3650.
- Schröder J, Schüller V, May A, Gerges C, Anders M. et al. Identification of loci of functional relevance to Barrett’s esophagus and esophageal adenocarcinoma: Cross-referencing of expression quantitative trait loci data from disease-relevant tissues with genetic association data. PLoS ONE, (2019); 14: e0227072.
- Shao Y, Guo X, Zhao L, Shen Y, Niu C. et al. A Functional Variant of the miR-15 Family Is Associated with a Decreased Risk of Esophageal Squamous Cell Carcinoma. DNA and Cell Biology, (2020); 39:1583–1594.
- Cui Q, Peng L, Wei L, Chang J, Tan W. et al. Genetic variant repressing ADH1A expression confers susceptibility to esophageal squamous-cell carcinoma. Cancer Letters, (2018); 421:43–50.
- Peng L, Cheng S, Lin Y, Cui Q, Luo Y. et al. CCGD-ESCC: A Comprehensive Database for Genetic Variants Associated with Esophageal Squamous Cell Carcinoma in Chinese Population. Genomics, Proteomics & Bioinformatics, (2018); 16:262–268.
- Agrawal N, Jiao Y, Bettegowda C, Hutfless SM, Wang Y. et al. Comparative Genomic Analysis of Esophageal Adenocarcinoma and Squamous Cell Carcinoma. Cancer Discovery, (2012); 2:899–905.
- Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, et al. Genetic landscape of esophageal squamous cell carcinoma. Nature Genetics, (2014); 46:1097–1102.
- Song Y, Li L, Ou Y, Gao Z, Li E. et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature, (2014); 509:91–95.
- Sawada G, Niida A, Uchi R, Hirata H, Shimamura T. et al. Genomic Landscape of Esophageal Squamous Cell Carcinoma in a Japanese Population. Gastroenterology, (2016); 150:1171–1182.
- Cui Y, Chen H, Xi R, Cui H, Zhao Y. et al. Whole-genome sequencing of 508 patients identifies key molecular features associated with poor prognosis in esophageal squamous cell carcinoma. Cell Research, (2020); 30:902–913.
- Cao W, Lee H, Wu W, Zaman A, McCorkle S. et al. Multi-faceted epigenetic dysregulation of gene expression promotes esophageal squamous cell carcinoma. Nature Communication, (2020); 11:3675.
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of oesophageal carcinoma. Nature (2017); 541:169–175.
- Zhang L, Zhou Y, Cheng C, Cui H, Cheng L. et al. Genomic Analyses Reveal Mutational Signatures and Frequently Altered Genes in Esophageal Squamous Cell Carcinoma. The American Journal of Human Genetics, (2020); 107:375.
- Yan T, Cui H, Zhou Y, Yang B, Kong P, et al. Multi-region sequencing unveils novel actionable targets and spatial heterogeneity in esophageal squamous cell carcinoma. Nature Communication, (2019); 10:1670.
- Cheng C, Zhou Y, Li H, Xiong T, Li S. et al. Whole-Genome Sequencing Reveals Diverse Models of Structural Variations in Esophageal Squamous Cell Carcinoma. The American Journal of Human Genetics, (2016); 98:256–274.
- Chen XX, Zhong Q, Liu Y, Yan SM, Chen ZH. et al. Genomic comparison of esophageal squamous cell carcinoma and its precursor lesions by multi-region whole-exome sequencing. Nature Communication, (2017); 8:524.
- Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nature Genetics, (2014); 46:467–473.
- Lin DC, Dinh HQ, Xie JJ, Mayakonda A, Silva TC, et al. Identification of distinct mutational patterns and new driver genes in oesophageal squamous cell carcinomas and adenocarcinomas. Gut, (2018); 67:1769–1779.
- Qin H-D, Liao X-Y, Chen Y-B, Huang S-Y, Xue W-Q. et al. Genomic Characterization of Esophageal Squamous Cell Carcinoma Reveals Critical Genes Underlying Tumorigenesis and Poor Prognosis. American Journal of Human Genetics, (2016); 98:709–727.
- Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F. et al. Somatic mutant clones colonize the human esophagus with age. Science, (2018); 362:911–917.
- Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H. et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature, (2019); 565:312–317.
- Chang J, Tan W, Ling Z, Xi R, Shao M. et al. Genomic analysis of oesophageal squamous-cell carcinoma identifies alcohol drinking-related mutation signature and genomic alterations. Nature Communication, (2017); 8:15290.
- Dong J, Maj C, Tsavachidis S, Ostrom QT, Gharahkhani P. et al. Sex-Specific Genetic Associations for Barrett’s Esophagus and Esophageal Adenocarcinoma. Gastroenterology, (2020); 159:2065-2076.e1.
- Deng J, Chen H, Zhou D, Zhang J, Chen Y, et al. Comparative genomic analysis of esophageal squamous cell carcinoma between Asian and Caucasian patient populations. Nature Communication, (2017); 8:1533.
- Hao JJ, Lin DC, Dinh HQ, Mayakonda A, Jiang Y-Y. et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nature Genetics, (2016); 48:1500–1507.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0