

Full Length Research Article
Gene Expression Profiling to Predict Prognostic Biomarkers for Relapse in Multiple Myeloma
Sana Elahi*, Sahar Fazal
Adv. life sci., vol. 11, no. 2, pp. 329-337, May 2024
*– Corresponding Author: Sana Elahi (sana_ilahi@yahoo.com)
Authors' Affiliations
[Date Received: 18/06/2023; Date Revised: 09/01/2024; Date Available Online: 18/04/2024]
Editorial Note on Version of Record
29 May 2025: This article has been corrected. See https://doi.org/10.62940/als.v13i0.4279 for more information.
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Multiple myeloma (MM), an incurable malignancy of plasma cells (PCs), is the second most common hematological cancer caused primarily by structural variations (chromosomal aberrations and somatic mutations). The majority of the patients acquired resistance against standard therapeutic approaches for MM and experienced relapse despite the continuous advances in MM therapies.
Methods: We performed Differential Gene Expression (DGE), literature mining and SNV analysis of Next Generation Sequencing (NGS) data of Newly Diagnosed MM (NDMM) and Relapsed/Refractory MM (RRMM). The selected Differentially Expressed Genes (DEGs) were subjected to functional enrichment and pathway analysis. Immune cells infiltration analysis was also performed to estimate immune cells variations in the Tumor Microenvironment (TME) of RRMM.
Result: CSF1R, VCAN, NRP1, COL22A1, BPI, BIRC5, MNX1, FAT1, ERG, TCL1A, AFF3 were selected after DGE, literature mining and SNV analysis. The functional enrichment of these DEGs showed significant enrichment for positive regulation of cell population proliferation, serene/threonine kinase activity, endothelial cell proliferation, cytokine binding, G protein activity and GDP binding, whereas KEGG pathway analysis revealed vital role of PI3K-Akt signaling pathway along with various cancer pathways. The immune cells infiltration analysis revealed the higher count of neutrophils and lesser level of T cells (CD8+) in TME of RRMM.
Conclusion: Our study suggests that neutrophils play an important role in modulation of TME in RRMM. The selected DEGs have previously been identified in progression, drug resistance and relapse of various cancers. The role of these biomarkers in RRMM has not been explored yet. Therefore, neutrophils, selected DEGs and PI3K-Akt signaling pathway are potential targets to investigate in RRMM.
Keywords: Multiple Myeloma; Relapse; Differential Expression; Immune Cell Infiltration; Literature Mining
Introduction![]()
MM is an incurable neoplastic malignancy of the fully differentiated B cells, PCs, which is characterized by aberrant division of PCs in Bone Marrow (BM) [1]. The uncontrolled division of monoclonal PCs leads to abnormal accumulation of non-functional immunoglobulins that result in anemia, hypercalcemia, renal dysfunction and lytic bone lesions [2]. MM is the second most prevalent hematological cancer, accounting for approximately 1% and 10% of all cancers and overall hematological neoplasms, respectively [3]. MM follows a multi-step process of pathogenesis initiating from precursor disorders i.e. Monoclonal Gammopathy of Undetermined Significance (MGUS) and smoldering myeloma that progresses to MM [2]. Chromosomal translocations of the IGH gene (14q32) reported in up to 50% of MM patients are most common and primary genomic events, which alter the expression of five oncogenes including CD1, FGFR3, CCND3, MAFB and MAF whereas MYC affecting translocations are reported in 15-20% newly diagnosed cases of MM [4]. Common secondary genomic events are Copy Number Variations (CNV) (hyperdiploidy, chromosome-arm events, loss of chromosome 13 and miscellaneous chromosomal gains & losses) that can contribute significantly to MM progression by promoting genomic instability [5]. Previously, no mutational land scape has been specified for MM compared to other hematological disorders, despite the fact that 60 out of 250 recurrently mutated genes were found as driver genes in MM according to the Whole Genome Sequencing (WGS) and Whole Exome Sequencing (WXS) studies. KRAS, TP53, NRAS, DIS3, BRAF, FAM46C, TRAF3, EGR1, ROBO1, FAT3 and SP140 were presented as the most frequently mutated genes [6]. Many researchers suggested that the dysregulation of gene expression has been associated with the dysregulation of cancer pathways [7]. These mutated genes are involved in different pathways such as MAPK pathway, DNA repair pathway, NF-kB pathway, RNA processing pathway, cell migration, adhesion, regulation of neurons, cell-cycle control pathway, B cell differentiation pathway, JAK-STAT pathway and PI3K pathway, most of which have been found dysregulated in MM [5]. Moreover, immune system of many MM patients has been observed dysfunctional i.e. low expression of tumor antigens and Human Leukocyte Antigen (HLA), presence of regulatory T cells (Tregs) and Myeloid-Derived Suppressor Cells (MDSCs), and enhanced expression of Programmed Cell Death Ligand 1 (PD-L1) [8]. Considering the crucial role of the immune system in MM, quantitative elucidation of the infiltrating immune cells in the TME can contribute significantly to the cancer treatment as it provides the information regarding the extent of immune evasion as well as cancer growth and progression [9].
The standard treatment options available for MM are combinatorial use of different classes of drugs, like proteasome inhibitors (bortezomib, carfilzomib and ixazomib), immunomodulatory drugs (thalidomide, lenalidomide, pomalidomide), alkylating agents, monoclonal antibodies (anti-cd38, elotuzumab), histone deacetylase inhibitors (panobinostat, vorinostat), corticosteroids, anthracyclines and autologous stem cells transplantation [3]. Besides the availability of vast treatment options, the 5- year survival rate has been observed only in 54% patients and majority of the MM patients experience relapse eventually. The ultimate cause for the relapse in MM is still unknown, however drug resistance is believed to be one of the major causes [3]. The underlying mechanisms of cancer progression and drug resistance are still not understood completely as drug resistance itself is governed by multiple factors including genetic and epigenetic variations, abnormal drug transport and metabolism, persistence of cancer stem cells, dysfunctional TME, immunotherapy antigens and dysregulation of apoptosis. However, with the advancement in genomic technologies and integrated approaches, several new treatment approaches are being investigated that can be promising against cancer treatment [10]. The role of genetic heterogeneity in the development of resistance against all available treatments and relapse is very little known in the case of RRMM as compared to the NDMM. Previously, relapse specific DEGs, dysregulated proteins and drug resistance have been reported in acute myeloid leukemia (AML) employing RNA-seq, WGS and WES analysis [11]. Thus, these genomic and transcriptomics-based methods can be useful to reveal novel biomarkers involved in acquisition of drug resistance and relapse in MM as well as the identification of novel onco-targets for drug development [12].
This study aims at utilizing RNA-Seq, WXS and WGS data of “Clinical Outcomes in Multiple Myeloma to Personal Assessment of Genetic Profile” (CoMMpass) (https://portal.gdc.cancer.gov/projects) of Multiple Myeloma Research Foundation (MMRF) to identify novel biomarkers involved in the pathogenesis and aggressiveness of MM. These biomarkers were further investigated for their association with dysregulation of the biological pathways and TME that can be involved in progression, drug resistance and relapse in RRMM.
Methods![]()
Data Collection
This study utilized the WXS, WGS and RNA-Seq data of NDMM and RRMM patients. The RNA-Seq and variant data was retrieved from National Cancer Institute Genomic Data Commons (NCI-GDC) data Portal utilizing the MMRF CoMMpass study data (https://portal.gdc.cancer.gov/projects.) GDC is a central repository, publicly available to access the vast amount of clinical and genomic data of numerous cancer studies. The MMRF CoMMpass dataset consists of WXS, WGS, RNA-Seq and clinical data of 995 MM patients primarily at the diagnosis and then at the relapse of cancer .
Shortlisting of Candidate Relapse Biomarkers
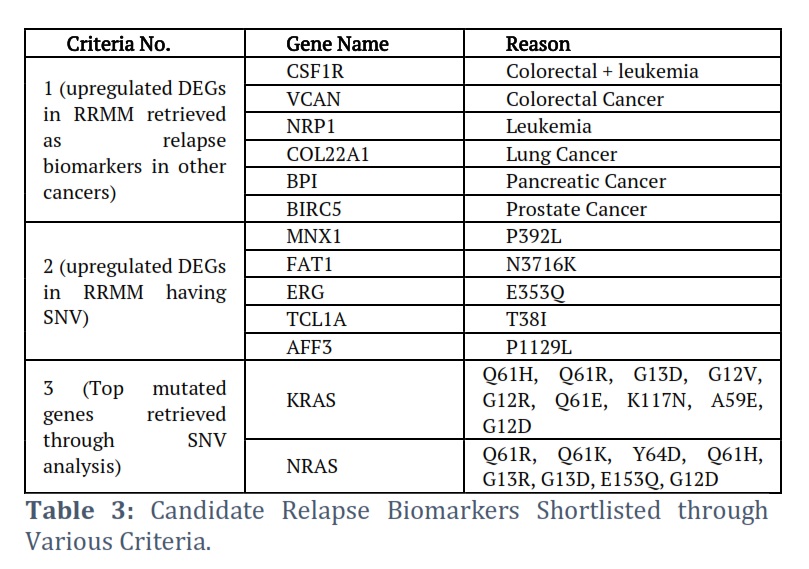
The genes were selected as candidate biomarkers if meeting any one of the following criteria 1) a gene retrieved as relapse biomarker in literature mining and also upregulated in DGE, 2) a gene carrying SNV and also upregulated in DGE, 3) Frequently mutated genes with higher no of SNVs.
Immune Cells Infiltration
The immune cells infiltration of the RRMM vs NDMM patients was carried out by utilizing the immunedeconv 2.1.0 package in R 4.2.2 [13]. This method estimates the count of immune cells present in the samples by employing deconvolute functions. The gene expression matrix was normalized to obtain fragments per kilobase of transcript per million mapped reads (FPKM) using the count2FPKM function of RNAAgeCalc 1.10.0 package [14]. The FPKM normalized matrix was further subjected to deconvolution methods to perform immune cells infiltration. Here we utilized two deconvolution methods i.e., quanTIseq and MCPcounter, whereas the ggplot2 version 3.4.1 was used to visualize the results in the form of bar chart and dot plot [13,15]. The quanTIseq utilizes bulk RNA-seq data to estimate fraction of 10 immune cell types along with the uncharacterized cells. Whereas MCPcounter obtains transcriptomics data to provides the absolute count of immune and stromal cells across all samples with respect to each cell type individually.
Literature Mining
Literature mining was conducted to identify biomarkers involved in recurrence of multiple types of cancers using the search terminologies on Google Scholar (https://scholar.google.com) and PubMed (https://pubmed.ncbi.nlm.nih.gov) as “recurrence in cancer”, “cancer relapse, “recurrent genes involved in lung cancer”, “recurrent genes involved in pancreatic cancer”, “recurrent genes involved in bladder cancer”, “recurrent genes involved in breast cancer”, “recurrent genes involved in colorectal cancer” and “recurrent genes involved in prostate cancer”. Furthermore, genes associated with relapse of MM were also identified through literature review using the terminologies “Relapse” “RRMM”, “cancer relapse” and “multiple myeloma” in Google Scholar and PubMed. Lastly the common genes obtained through literature mining and DGE was identified using grep tool on Linux.
Differential Gene Expression
Differential gene expression of RNA-seq data of CoMMpass was performed by using DESeq2 (1.38.3), an R (4.2.2) package [16]. This method normalizes the counts of each gene and employs shrinkage estimation resulting in more stable and explainable calculations focused on strength based quantitative analysis [16]. The DEGs were selected on the basis of log2 fold change (Log2FC) and p-value. Log2FC and p-value were set at the threshold of 2.0 < Log2FC < -2.0 and < 0.05, respectively to filter out up and downregulated DEGs. Furthermore, the EnhancedVolcano 1.16.0 package was used to retrieve EnhancedVolcano plot of DEGs [17].
Function Enrichment
Functional enrichment analysis of DEGs and shortlisted candidate biomarkers was performed by GeneCodis 4 (https://genecodis.genyo.es/). GeneCodis 4 is an online enrichment analysis tool that extracts biological information from the experimental data by giving the statistical score for significantly enriched annotations [18].The lists of upregulated and downregulated genes were taken separately as input for annotations specified as Gene Ontology (GO) terms (biological process (BP), cellular component (CC), molecular function (MF)) and KEGG pathway.
Results![]()
After applying the filter of “BM samples” and “non-synonymous SNV” on MMRF- CoMMpass dataset 833 patients were retrieved. Among them 753 were NDMM patients containing 2007 SNVs in 454 genes whereas 80 were RRMM patients along with 226 SNVs affecting 152 genes. The top 5 Missense SNVs maximum affecting cases in cohort of NDMM and RRMM were Q61R in NRAS (7.56%), Q61H in KRAS (6.72%), Q61K in NRAS (5.40%), V640E in BRAF (3.48%) and G12D in KRAS (3.36%).
Identification of Immune Cells Fraction in MM Samples
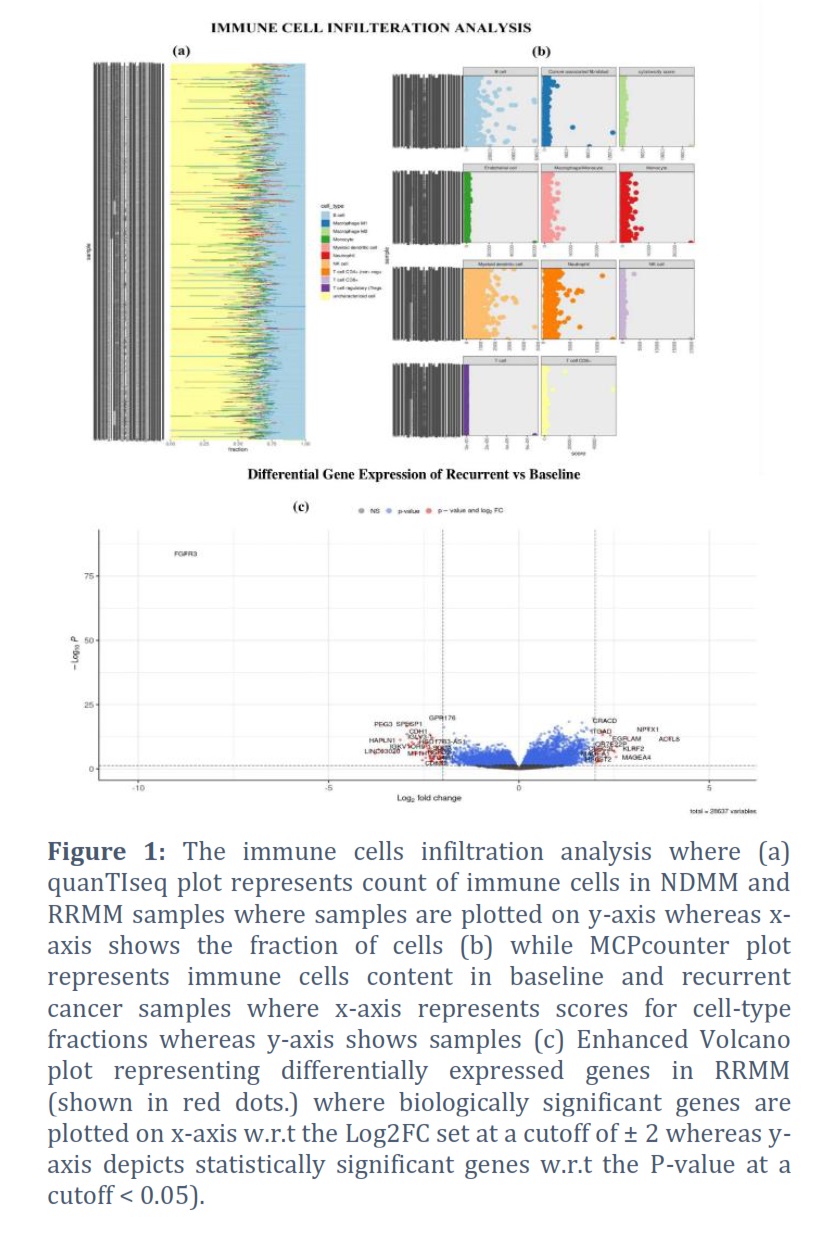
The results of immune cell infiltration analysis were assembled in Figure 1a and b. The meticulous analysis of results revealed that, T cell (CD4), natural killer cells (NK), monocytes, macrophages and myeloid dendritic cells (MDC) counts was marginally raised for RRMM in comparison to NDMM. Cytotoxicity score, endothelial cells and cancer associated fibroblasts (CAFs) can only be calculated through MCPcounter showed the similar trend of slightly higher count in RRMM as compared to NDMM. Similarly, regulatory T cell (Tregs) can only be estimated by quanTIseq was somewhat more in RRMM than NDMM. The only discrepancy was observed in the count of B cells as it was more at RRMM than NDMM according to MCPcounter whereas it was higher in NDMM than RRMM according to quanTIseq. Moreover, T cell (CD8+) count declined, and Neutrophils count amplified in RRMM in comparison to NDMM according to both methods (Figure 1a and b).
DGE Analysis
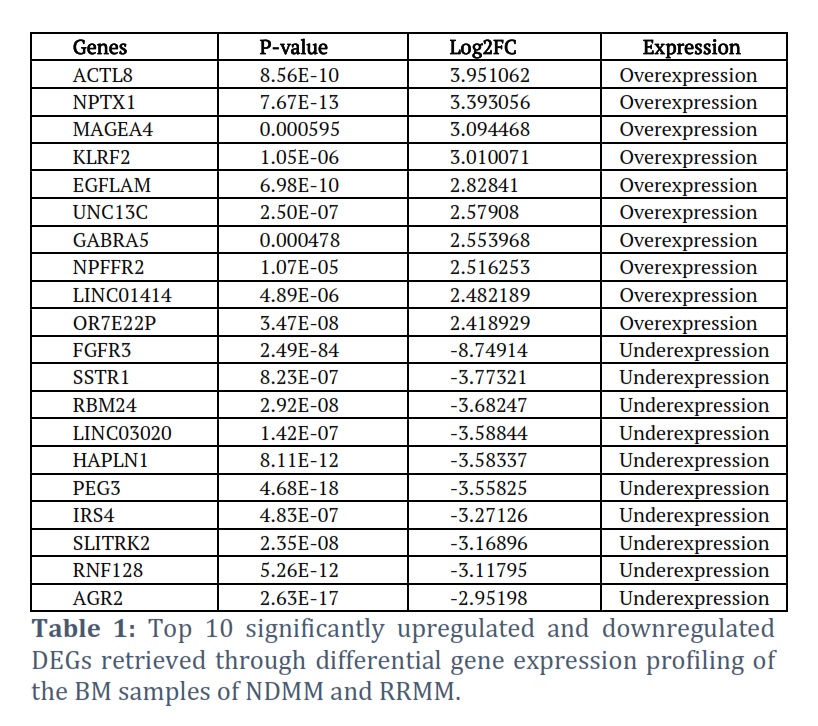
The gene expression profiling of the BM samples of NDMM and RRMM patients revealed a total of 1562 dysregulated genes. Among them 908 genes were significantly upregulated in RRMM (Log2FC > 2, p-value < 0.05) whereas 654 genes were downregulated (Log2FC < -2, p-value < 0.05) as shown in Figure 1c. Furthermore, the top 10 upregulated and downregulated genes were filtered out on the basis of highest and lowest Log2FC values are assembled in Table 1. The top 10 significantly upregulated DEGs include ACTL8, NPTX1, MAGEA4, KLRF2, EGFLAM, UNC13C, GABRA5, NPFFR2, LINC01414, OR7E22P. Whereas the top10 significantly downregulated DEGs were FGFR3, SSTR1, RBM24, LINC03020, HAPLN1, PEG3, IRS4, SLITRK2, RNF128, AGR2.
Literature Mining
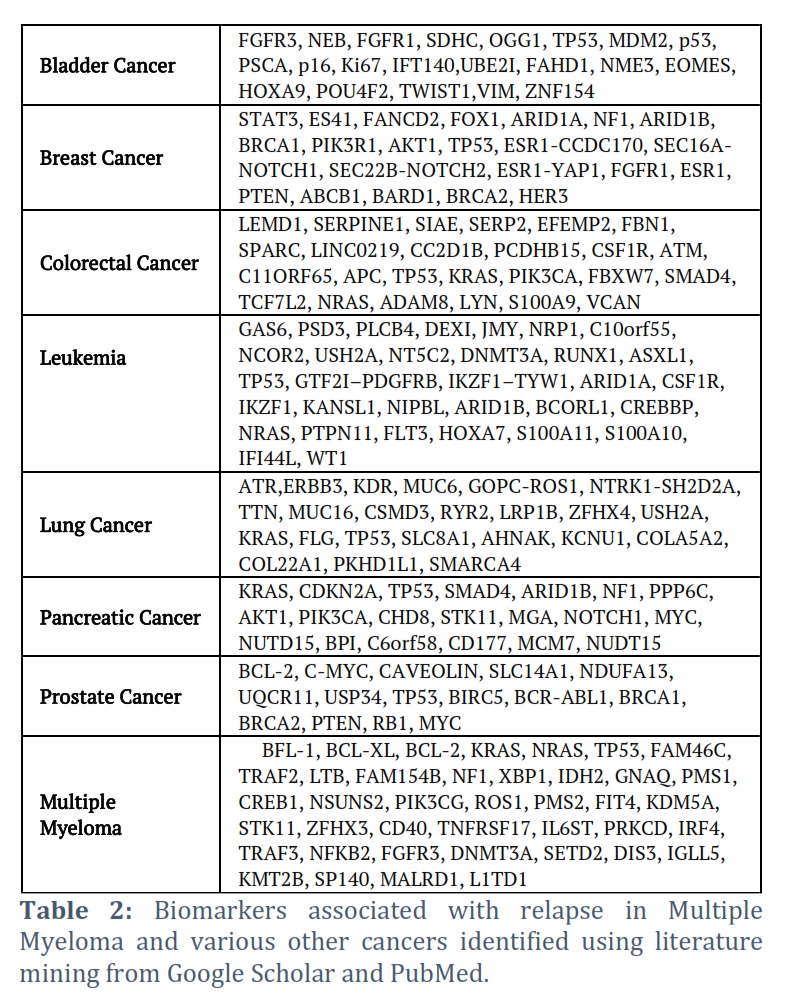
Literature mining retrieved 136 genes with significant role in relapse of various cancers as shown in Table 2. Among them 40 genes were found to be involved in RRMM (Table 2). Analysis of results revealed that KRAS, NRAS, TP53, NF1, STK11 and DNMT3A that were found as relapse biomarker in MM also reported for relapse in other cancers. Although these biomarkers were not found to be upregulated in our DGE analysis but mutations in these gene is reported by various research in RRMM.
Shortlisting of Candidate Genes
Genes were shortlisted on the basis of following criteria 1) a gene retrieved as relapse biomarker in literature mining and also upregulated in DGE, 2) a gene carrying SNV and also upregulated in DGE, 3) Top mutated genes retrieved through SNV analysis. According to first criteria 6 DEGs were shortlisted including Colony-Stimulating Factor-1 Receptor (CSF1R), VCAN, Neuropilin 1(NRP1), Collagen Type XXII Alpha 1 Chain (COL22A1), Bactericidal Permeability Increasing Protein (BPI) and Baculoviral inhibitor of apoptosis repeat-containing 5 (BIRC5). These DEGs were upregulated in our analysis but reported as relapse biomarkers for other cancers (Table 3) in literature. Similarly, 5 DEGs were selected in accordance with second criteria included Motor Neuron and Pancreas Homeobox 1 (MNX1), FAT Atypical Cadherin 1 (FAT1) ETS Transcription Factor ERG (ERG), TCL1 Family AKT Coactivator A (TCL1A) and ALF Transcription Elongation Factor 3 (AFF3) (Table 3). These DEGs showed upregulation along with SNV in RRMM patients. These DEGs are reported as diagnostics, prognostics, therapeutic and relapse biomarkers for various other cancers but their role in relapse of MM needs to be explored. Moreover, two genes (KRAS, NRAS) were selected according to third criteria although, didn’t show any significant DE at relapse but harbor multiple SNVs in both NDMM and RRMM patients (Table 3).
Functional Enrichment and Pathway Analysis of DEGs and Shortlisted Candidate Relapse Biomarker
The GO term analysis of DEGs revealed that upregulated DEGs were found to be significantly enriched in the following BP, immune response, immune system process, inflammatory response, cell adhesion, positive regulation of T cell activation, antigen processing and presentation of peptides, peptide antigen assembly with MHC class II, immunoglobulin production, antigen processing and presentation of exogenous peptides and neutrophil chemotaxis. However, the downregulated DEGs were notably associated with adaptive immune response, positive regulation of B cell activation, phagocytosis recognition and engulfment, complement activation and development of central nervous system among many others. Similarly, the significant CC terms for upregulated DEGs were plasma membrane, extracellular region and extracellular space whereas, collagen-containing extracellular matrix, external side of plasma membrane, tertiary granule membrane, cell surface, ficolin-1-rich granule lumen and MHC class II protein complex were also notable terminologies. However, the downregulated DEGs were also enriched in plasma membrane, extracellular region and extracellular space, collagen-containing extracellular matrix, external side of plasma membrane along with immunoglobulin complex, synapses, and glutamatergic synapse. Additionally, the significant MF retrieved for the upregulated DEGs through GO analysis were carbohydrate binding, signaling receptor activity, transmembrane signaling receptor activity and cytokine activity. While the antigen binding, sequence-specific DNA binding was the most significant MF related to downregulated DEGs among many others.
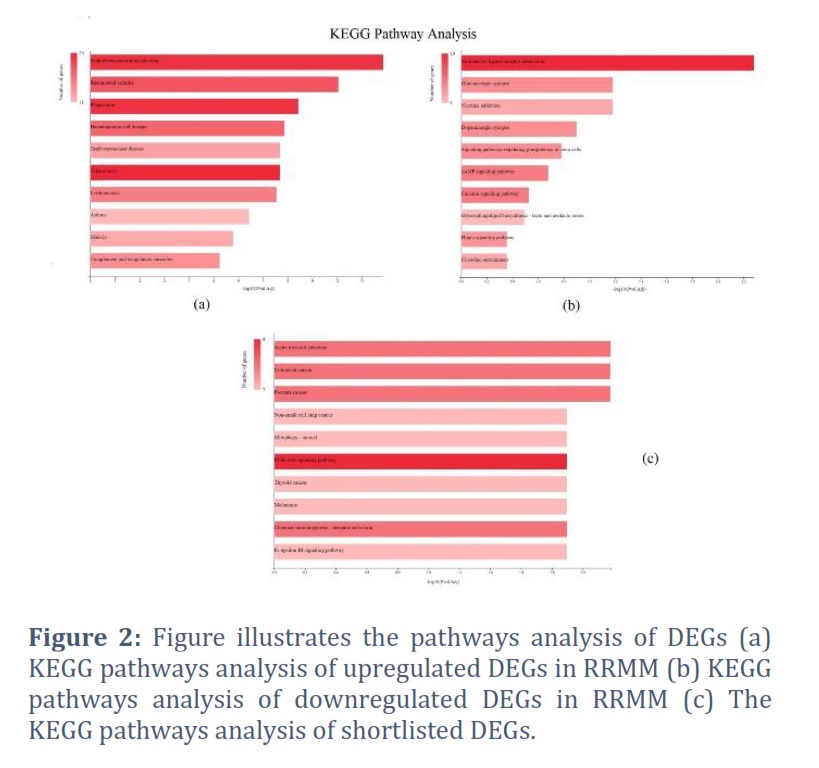
The pathway analysis of DEGs revealed their enrichment in various essential pathways (Figure 2). The upregulated DEGs were found significantly enriched in numerous disease pathways including staphylococcus aureus infection, rheumatoid arthritis, malaria, Tuberculosis, asthma, leishmaniasis, graft versus host disease along with phagosome and hematopoietic cell lineage (Figure 2a). The downregulated DEGs, on the other hand, were found to be enriched in diverse signaling pathways including cAMP signaling, calcium signaling, hippo signaling, signaling pathways regulating pluripotency of stem cells along with Neuroactive ligand-receptor interaction and various synapses as depicted in Figure 2b.
Furthermore, the functional enrichment of these shortlisted candidate genes showed significant enrichment of the following BP ontologies, positive regulation of cell population proliferation, multicellular organism development, positive regulation of serene/threonine kinase activity and positive regulation of endothelial cell proliferation more significant among many others. The shortlisted DEGs showed CC enrichment more significantly in focal adhesion whereas, G protein activity, GDP-binding and cytokine binding were enriched MF. Moreover, KEGG pathways analysis revealed significant enrichment in prostate cancer, colorectal cancer (CRC), acute myeloid leukemia, chemical carcinogenesis receptor activation and PI3K/AKT signaling pathway (Figure 2c).
Figures & Tables
The relapse in MM is a constant concern for researchers because of complex multifactorial onset, prognosis and even more intricate recurrence of cancer. Development of effective treatment regimen to minimize relapse and maximize progression free survival of MM patients is conditional with the hunt of new biomarkers for diagnosis, prognosis and relapse along with efficient and precise drug targets and compounds. Literature review of six genes (CSF1R, VCAN, NRP1, COL22A1, BPI and BIRC5) identified by combine analysis of literature mining and DGE revealed their potential in modulating TME and disease progression. CSF1R, a receptor tyrosine kinase, inhibition is reported as potential antitumor strategy. Activated CSF1R are recruited by tumor-associated macrophages and release cytokine that modulated TME to protumoral phenotype. CSF1R activation also initiates many downstream pro survivals signaling cascades including PI3K/AKT, ERK1/2, and JNK [19]. Similarly, BIRC5 is also an immune-related gene that inhibits apoptosis and promotes cellular proliferation. High expression of BIRC5 regulates DNA methylation hence is reported as potential target for developing immunotherapies [20]. Moreover, NRP1 is an independent predictor of relapse and poor survival in non-small cell lung cancer (NSCLC). It is also reported as novel potential therapeutic target in NSCLC because of its critical role in tumorigenesis, cancer invasion, and angiogenesis through VEGF, PI3K, and Akt pathways [21]. Whereas VCAN and COL22A1 are reported as prognostic biomarkers in various cancers. VCAN mRNAs are specifically expressed in cancer-associated fibroblasts and associated with poor relapse free survival of stage II-III patients in CRC. It is a promising biomarker to identify stage II-III patients with high risk of relapse in CRC [22]. While COL22A1 is an integral part of novel prognostic immune related gene signature in CRC [23]. Additionally, COL22A1 is also reported as poor prognosis and relapse biomarker in Head and neck squamous cell carcinoma (HNSCC) [24]. Furthermore, BPI is associated with human neutrophil as in response to inflammation neutrophils secrets BPI along with many other cytotoxic proteins and can promote tumor metastasis by the formation of so-called neutrophil extracellular traps (NETs).
High levels of circulating and intratumoral neutrophils have been shown to correlate with poor survival in pancreatic cancer and pancreatic ductal adenocarcinoma [25]. These results were also consistent with our immune cell infiltration analysis which revealed the presences of higher neutrophils count in RRMM. The immune cells infiltration analysis in our study corroborated the previously reported role of immune dysfunction in MM invasion and progression [19]. Development of effective therapeutic strategies against neutrophils for treating cancer and various other diseases has already been in consideration. According to recent studies neutrophils have multiple phenotypes that perform diverse functions, particularly modulation of inflammation and immune response [26]. Similarly, another study mentioned the impairment of immune response in MM due to reduced phagocytic activity of neutrophils in comparison to healthy control [27]. Moreover, the role of neutrophils in facilitating tumor progression through immune deregulation and increased vulnerability to infection in MUGS and MM has already been established [28]. CD8+ T cells count was found to be decreased in NDMM, are imperative for immune defense against intracellular pathogens (viruses, bacteria) and for tumor surveillance. The trafficking or transporting of CD8+ T cells into the TME is crucial to exert its anti-tumor function. The elevated levels of CD8+ T cells in the TME are linked with positive anti-tumor effects and good prognosis in breast, colorectal, glioblastoma, and cervical cancers [29]. Hence the lower count of CD8+ T in TME is not only linked with cancer progression/relapse or poor prognosis but also with the increased risk of secondary viral or bacterial infection. The lower count of CD8+ T cells retrieved through immune infiltration investigation also supports our findings of pathway analysis which showed enrichment of various other cancer and bacterial infections pathways in RRMM (Figure 2).
The DGE and SNV analysis of dataset retrieved five genes (MNX1, FAT1, ERG, TCL1A, AFF3) harboring mutation and upregulation in RRMM. These mutations include MNX1 (P392L), FAT1 (N3716K), ERG (E353Q), TCL1A (T38I), and AFF3 (P1129L). KRAS and NRAS, although not found in DGE but are the most mutated genes in both NDMM and RRMM patients according to SNV data. Both KRAS and NRAS are proto-onco genes, belonging to the Ras family of proteins, small GTPase, involved in regulation of biological processes particularly cell growth, proliferation and apoptosis [30]. It has been reported that newly acquired mutations and pre-treatment sub-clonal mutations of KRAS and NRAS in MM possibly induce chemo resistance and relapse [31]. Mutation in KRAS and NRAS have been reported to be more common in relapsed patients (>70% patients) [32,33]. The codons G12, Q61 and G13 are mutation hotspots for both KRAS and NRAS but various substitutions in each codon elicit different signaling pathway, hence express distinct pathophysiology [34]. In our study all the three hotspots were found substituted with various codon (NRAS: Q61H, Q61R, Q61E, G12V, G12D, G12R, G13D KRAS: Q61K, Q61H, G12D, G13R, G13D) along with few new codons (NRAS: K117N, A59E KRAS: Y64D, E153Q) in RRMM. The impairment of PI3K pathway due to KRAS (G12R) mutation is reported in literature [35] and is also consistent with our results of pathway analysis (Figure 4). Additionally, MNX1 encodes a transcription factor (HB9) that contains a homeodomain. The overexpression of MNX1 has been reported in many cancers (prostate, colon, liver, breast and bladder cancer, glioma and pancreatic progenitor tumors and acute myeloid leukemia) and has been suggested as potential diagnostic and prognostic biomarker [36, 37]. Upregulation of MNX1 stimulates the Wnt/β‐catenin signaling and via expression of downstream genes c‐Myc and CCND1, hence plays a vital role in CRC progression [38]. FAT1 is among the most frequently mutated genes in many types of cancer. The role of FAT1 in cancer progression is highly dependent upon cancer type. In some cancers epithelial-mesenchymal transition (EMT) and the formation of cancer initiation/stem-like cells is promoted by loss of FAT1 function promotes whereas overexpression of FAT1 leads to EMT in others. The paired analysis of diagnosis and relapse sample in B-cell acute lymphoblastic leukemia overexpression of FAT1 was found correlated with shorter relapse-free and overall survival [39]. Several studies have reported a correlation of FAT1 mutation or expression with prognosis in various cancers, such as breast cancer, NSCLC, gastric cancer and T-cell lymphoma [40]. Positive correlation of FAT1 overexpression with proliferation and WNT/β‐catenin signaling pathway in T-cell acute leukemia (T-ALL) is recently reported in a study [41]. ERG encodes a transcription factor involved in development and differentiation affecting vasculogenesis, haematopoiesis, angiogenesis and embryogenesis, and is associated with regulation of cellular processes [42]. ERG over expression is associated with poor prognosis and oncogenesis promotion, in prostate cancer, Ewing’s sarcoma, acute myeloid leukemia, acute T-lymphoblastic leukemia [43]. ERG high expression stimulates gene fusion event (ERG-TMPRSS2) that leads to early relapse in AML [43]. ERG upregulation is associated with upregulation of PI3K/AKT pathway [44]. TCL1A is a proto-oncogene expressed in embryonic stem cells, activated T and B lymphocytes and coactivator of kinases and interacting partners crucial in signaling pathways (PI3K and NF-κB) and cellular activities [45]. Dysregulated TCL1A has a well-documented role in hematopoietic malignancies i.e., development of T-cell leukemia, correlation of overexpression with aggressiveness, deregulation of the cell cycle and genomic instability in chronic lymphocytic leukemia [46]. It is also suggested as prognostic biomarker for stage II/III CRC [47], therefore, proposed as potential biomarker for colorectal and hematological malignancies [45, 47]. AFF3 is primarily expressed in B cells and encodes a protein involved in transcription regulation [48]. AFF3 upregulation has been found in many cancers (gastric, breast, Adrenocortical and AML), also involved in modulating TME thus suggested as target for immune therapy [48-50]. In gastric cancer, dysregulated AFF3 is a potential marker for diagnosis and prognosis as well as correlated with immune checkpoints response whereas in breast cancer overexpression is associated with drug (tamoxifen) resistance therefore suggested as predictive marker for ER+ breast cancer [48, 50]. Moreover, AFF3 meditates oncogenic effects of β-catenin as constitutive activation of Wnt/β-catenin signaling pathway in mice leads to formation of malignant adrenocortical tumors [51].
The functional enrichment analysis of all shortlisted genes revealed their significant enrichment in G protein activity, GDP binding, positive regulation of cell population proliferation, serene/threonine kinase activity, endothelial cell proliferation. However, pathway enrichment for PI3K-Akt signaling and prostate cancer, acute myeloid leukemia and CRC pathways along with many cancer pathways was also significant (Figure 2). It can be inferred that KRAS and NRAS mutants may affect the activation and hydrolysis due to dysregulation of G protein activity, GDP and GTP binding, and GTPase activity. The aberrant GTPase activity due to KRAS mutations has been reported to affect the GTP-hydrolysis [52]. Furthermore, enrichment of PI3K-Akt signaling pathway was also consistent with our other results as upregulation of CSF1R, NRP1, TCL1A and ERG activate PI3K-Akt signaling [5, 44]. This pathway is crucial to many cellular processes and plays a significant role in cancer proliferation and multidrug resistance. Decrease in cellular apoptosis is mediated by continuous phosphorylation of various transcription factors by AKT, thus the promotion of the proliferation, angiogenesis, and survival of cell [53]. However, the PI3K-Akt signaling pathway is crucial for pathophysiology of MM and is associated with therapy resistance [54, 55]. Moreover, Wnt/β‐catenin signaling plays a dual and disease stage-specific role in the pathogenesis of MM. Wnt/β‐catenin pathway activation during MM disease progression is mediated through epigenetic silencing by antagonists which facilitates pathway activation and proliferation of MM cells [56]. The pathway although not found enriched in pathway analysis but the 3 DEGs (MNX1, AFF3, and FAT1) among the 5 selected candidate genes are found to have direct relationship in upregulation of wnt/ β‐catenin pathway [38, 51]. This study proposed that upregulation of the following genes (CSF1R, VCAN, NRP1, COL22A1, BPI, BIRC5, MNX1, FAT1, ERG, TCL1A, AFF3) might be the cause of drug resistance and relapse in RRMM. These genes exert their effect by modulating apoptosis, TME and PI3K-Akt signaling pathway mainly along with various other pathways. Their role as potential biomarker for diagnosis, prognosis and drug resistance in various cancers has also been reported. Thorough investigation on role of these genes in drug resistance might be helpful in understanding the mechanism of relapse in MM. Furthermore, the neutrophils, P13K-Akt signaling pathway along with aforementioned genes are potential biomarkers of relapse in MM and targets of new effective treatment regimens.
Conflict of Interest
The authors declare that there is no conflict of interest.
All authors contribute equally to designing study, acquisition of data, analysis and interpretation of data, and drafting of the manuscript.
![]() References
References
- Palumbo A, Anderson K. Multiple Myeloma. The New England journal of medicine, (2011); 364(11): 1046-1060.
- Robert A Kyle MAG, Thomas E Witzig, John A Lust, Martha Q Lacy, Angela Dispenzieri, Rafael Fonseca, S Vincent Rajkumar, Janice R Offord, Dirk R Larson, Matthew E Plevak, Terry M Therneau, Philip R Greipp. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clinic Proceeding, (2003); 78(1): 21-33.
- Rajkumar SV. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. American journal of hematology, (2022); 97(8): 1086-1107.
- Palumbo A, Avet-Loiseau H, Oliva S, Lokhorst HM, Goldschmidt H, et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, (2015); 33(26): 2863-2869.
- Humaira Sadaf HH, Mohsin Maqbool, Kylin Emhoff, Jianhong Lin, Shan Yan, Faiz Anwer ,Jianjun Zhao. Multiple myeloma etiology and treatment. Journal of Translational Genetics and Genomics, ( 2022); 663-83.
- Maura F, Bolli N, Angelopoulos N, Dawson KJ, Leongamornlert D, et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nature communications, (2019); 10(1): 3835.
- Chaimaa Saadoune BN, Hasna Hamdaoui, Fatima Chegdani, and Faiza Bennis. Multiple Myeloma: Bioinformatic Analysis for Identification of Key Genes and Pathways. Bioinformatics and Biology Insights, (2022); 16: 11779322221115545.
- Nijhof IS, van de Donk N, Zweegman S, Lokhorst HM. Current and New Therapeutic Strategies for Relapsed and Refractory Multiple Myeloma: An Update. Drugs, (2018); 78(1): 19-37.
- Chen C, Li Y, Miao P, Xu Y, Xie Y, et al. Tumor immune cell infiltration score based model predicts prognosis in multiple myeloma. Scientific reports, (2022); 12(1): 17082.
- Pinto V, Bergantim R, Caires HR, Seca H, Guimaraes JE, et al. Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers, (2020); 12(2): 407.
- Stratmann S, Vesterlund M, Umer HM, Eshtad S, Skaftason A, et al. Proteogenomics analysis of acute myeloid leukemia associates relapsed disease with reprogrammed energy metabolism both in adults and children. Leukemia, (2023); 37(3): 550-559.
- Weaver CJ, Tariman JD. Multiple Myeloma Genomics: A Systematic Review. Seminars in oncology nursing, (2017); 33(3): 237-253.
- Sturm G, Finotello F, Petitprez F, Zhang JD, Baumbach J, et al. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics, (2019); 35(14): i436-i445.
- Ren X, Kuan PF. RNAAgeCalc: A multi-tissue transcriptional age calculator. PLoS One, (2020); 15(8): e0237006.
- Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, et al. Erratum to: Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome biology, (2016); 17(1): 249.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology, (2014); 15(12): 550.
- Kevin Blighe SR, Myles Lewis (2018) EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. https://github.com/kevinblighe/EnhancedVolcano.
- Garcia-Moreno A, Lopez-Dominguez R, Villatoro-Garcia JA, Ramirez-Mena A, Aparicio-Puerta E, et al. Functional Enrichment Analysis of Regulatory Elements. Biomedicines, (2022); 10(3).
- Wang J, Hu Y, Hamidi H, Dos Santos C, Zhang J, et al. Immune microenvironment characteristics in multiple myeloma progression from transcriptome profiling. Frontiers in Oncology, (2022); 12948548.
- Xu L, Yu W, Xiao H, Lin K. BIRC5 is a prognostic biomarker associated with tumor immune cell infiltration. Scientific reports, (2021); 11(1): 390.
- Hong TM, Chen YL, Wu YY, Yuan A, Chao YC, et al. Targeting neuropilin 1 as an antitumor strategy in lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research, (2007); 13(16): 4759-4768.
- Chida S, Okayama H, Noda M, Saito K, Nakajima T, et al. Stromal VCAN expression as a potential prognostic biomarker for disease recurrence in stage II-III colon cancer. Carcinogenesis, (2016); 37(9): 878-887.
- Wang Y, Li W, Jin X, Jiang X, Guo S, et al. Identification of prognostic immune-related gene signature associated with tumor microenvironment of colorectal cancer. BMC Cancer, (2021); 21(1): 905.
- Misawa K, Kanazawa T, Imai A, Endo S, Mochizuki D, et al. Prognostic value of type XXII and XXIV collagen mRNA expression in head and neck cancer patients. Molecular and clinical oncology, (2014); 2(2): 285-291.
- Deng M, Aberle MR, van Bijnen A, van der Kroft G, Lenaerts K, et al. Lipocalin-2 and neutrophil activation in pancreatic cancer cachexia. Frontiers in immunology, (2023); 14: 1159411.
- Rosales C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Frontiers in physiology, (2018); 9: 113.
- Parrinello N, Romano A, Conticello C, Cavalli M, La Fauci A, et al. Neutrophils Of Multiple Myeloma Are Dysfunctional and Immunosuppressive. Blood, (2013); 122(21): 3138-3138.
- Romano A, Parrinello NL, Simeon V, Puglisi F, La Cava P, et al. High-density neutrophils in MGUS and multiple myeloma are dysfunctional and immune-suppressive due to increased STAT3 downstream signaling. Scientific reports, (2020); 10(1): 1983.
- Maimela NR, Liu S, Zhang Y. Fates of CD8+ T cells in Tumor Microenvironment. Computational and structural biotechnology journal, (2019); 17: 1-13.
- Cox AD, Der CJ. Ras history: The saga continues. Small GTPases, (2010); 1(1): 2-27.
- Corre J, Cleynen A, Robiou du Pont S, Buisson L, Bolli N, et al. Multiple myeloma clonal evolution in homogeneously treated patients. Leukemia, (2018); 32(12): 2636-2647.
- Hu Y, Chen W, Wang J. Progress in the identification of gene mutations involved in multiple myeloma. OncoTargets and therapy, (2019); 12: 4075-4080.
- Sacco A, Federico C, Todoerti K, Ziccheddu B, Palermo V, et al. Specific targeting of the KRAS mutational landscape in myeloma as a tool to unveil the elicited antitumor activity. Blood, (2021); 138(18): 1705-1720.
- Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nature reviews. Drug discovery, (2014); 13(11): 828-851.
- Hobbs GA, Baker NM, Miermont AM, Thurman RD, Pierobon M, et al. Atypical KRAS(G12R) Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discovery, (2020); 10(1): 104-123.
- Ragusa D, Tosi S, Sisu C. Pan-Cancer Analysis Identifies MNX1 and Associated Antisense Transcripts as Biomarkers for Cancer. Cells, (2022); 11(22): 3577.
- Nilsson T, Waraky A, Ostlund A, Li S, Staffas A, et al. An induced pluripotent stem cell t(7;12)(q36;p13) acute myeloid leukemia model shows high expression of MNX1 and a block in differentiation of the erythroid and megakaryocytic lineages. International journal of cancer, (2022); 151(5): 770-782.
- Yang X, Pan Q, Lu Y, Jiang X, Zhang S, et al. MNX1 promotes cell proliferation and activates Wnt/beta-catenin signaling in colorectal cancer. Cell biology international, (2019); 43(4): 402-408.
- de Bock CE, Ardjmand A, Molloy TJ, Bone SM, Johnstone D, et al. The Fat1 cadherin is overexpressed and an independent prognostic factor for survival in paired diagnosis-relapse samples of precursor B-cell acute lymphoblastic leukemia. Leukemia, (2012); 26(5): 918-926.
- Chen ZG, Saba NF, Teng Y. The diverse functions of FAT1 in cancer progression: good, bad, or ugly? Journal of Experimental and Clinical Cancer Research, (2022); 41(1): 248.
- Liebig S, Neumann M, Silva P, Ortiz-Tanchez J, Schulze V, et al. FAT1 expression in T-cell acute lymphoblastic leukemia (T-ALL) modulates proliferation and WNT signaling. Scientific reports, (2023); 13(1): 972.
- Adamo P, Ladomery MR. The oncogene ERG: a key factor in prostate cancer. Oncogene, (2016); 35(4): 403-414.
- Khosh Kish E, Choudhry M, Gamallat Y, Buharideen SM, D D, et al. The Expression of Proto-Oncogene ETS-Related Gene (ERG) Plays a Central Role in the Oncogenic Mechanism Involved in the Development and Progression of Prostate Cancer. International journal of molecular sciences, (2022); 23(9): 4772.
- Zhou W, Su Y, Zhang Y, Han B, Liu H, et al. Endothelial Cells Promote Docetaxel Resistance of Prostate Cancer Cells by Inducing ERG Expression and Activating Akt/mTOR Signaling Pathway. Frontiers in oncology, (2020); 10: 584505.
- Paduano F, Gaudio E, Mensah AA, Pinton S, Bertoni F, et al. T-Cell Leukemia/Lymphoma 1 (TCL1): An Oncogene Regulating Multiple Signaling Pathways. Frontiers in oncology, (2018); 8: 317.
- Stachelscheid J, Jiang Q, Aszyk C, Warner K, Bley N, et al. The proto-oncogene TCL1A deregulates cell cycle and genomic stability in CLL. Blood, (2023); 141(12): 1425-1441.
- Li H, Yan X, Liu L, Huang L, Yin M, et al. T-cell leukemia/lymphoma-1A predicts the clinical outcome for patients with stage II/III colorectal cancer. Biomed Pharmacother, (2017); 88: 924-930.
- Shi Y, Zhao Y, Zhang Y, AiErken N, Shao N, et al. AFF3 upregulation mediates tamoxifen resistance in breast cancers. Journal of experimental & clinical cancer research, (2018); 37(1): 254.
- xi chen QY, Xiang Qin, Jing Liu, Yan Zeng, Sili Long, Wenjun Liu. High expression of AFFs is related to prognosis of acute myeloid leukemia. PREPRINT (Version 1) available at Research Square (2021); [https://doi.org/10.21203/rs.3.rs-927123/v1]
- Zeng Y, Zhang X, Li F, Wang Y, Wei M. AFF3 is a novel prognostic biomarker and a potential target for immunotherapy in gastric cancer. Journal of Clinical Laboratory Analysis, (2022); 36(6): e24437.
- Lefevre L, Omeiri H, Drougat L, Hantel C, Giraud M, et al. Combined transcriptome studies identify AFF3 as a mediator of the oncogenic effects of beta-catenin in adrenocortical carcinoma. Oncogenesis, (2015); 4(7): e161.
- Li C, Vides A, Kim D, Xue JY, Zhao Y, et al. The G protein signaling regulator RGS3 enhances the GTPase activity of KRAS. Science, (2021); 374(6564): 197-201.
- Shi YY, Meng XT, Xu YN, Tian XJ. Role of FOXO protein’s abnormal activation through PI3K/AKT pathway in platinum resistance of ovarian cancer. The journal of obstetrics and gynaecology research, (2021); 47(6): 1946-1957.
- Ramakrishnan V, Kumar S. PI3K/AKT/mTOR pathway in multiple myeloma: from basic biology to clinical promise. Leuk Lymphoma, (2018); 59(11): 2524-2534.
- Wang L, Lin N, Li Y. The PI3K/AKT signaling pathway regulates ABCG2 expression and confers resistance to chemotherapy in human multiple myeloma. Oncology Reports, (2019); 41(3): 1678-1690.
- van Andel H, Kocemba KA, Spaargaren M, Pals ST. Aberrant Wnt signaling in multiple myeloma: molecular mechanisms and targeting options. Leukemia, (2019); 33(5): 1063-1075.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0