

Full Length Research Article
Reconnoitering Mycobacterium tuberculosis lipoproteins to design subunit vaccine by immunoinformatics approach
Muhammad Ahsan Naeem#1*, Muhammad Muzammal Adeel#2, Ayesha Kanwal3, Sajjad Ahmad4, Waqas Ahmad1, Qaiser Akram1, Asif Saleh1 , Waqas Ahmed5
Adv. life sci., vol. 8, no. 3, pp. 300-306, July 2021
*- Corresponding Author: Muhammad Ahsan Naeem (Email: muhammadahsannaeem@ymail.com)
Authors' Affiliations
2. Agricultural Bioinformatics Key Laboratory of Hubei Province, Hubei Engineering Technology Research Center of Agricultural Big Data, College of Informatics, Huazhong Agricultural University, Wuhan – China
3. College of Life Sciences, University of Science and Technology of China, Hefei – People’s Republic of China
4. Department of health and biological sciences, Abasyn University Peshawar, Khyber Pakhtunkhwa – Pakistan.
5. Department of Biomedical and Diagnostic Sciences, University of Tennessee College of Veterinary Medicine, Knoxville, Tennessee – United States of America
#These authors equally contributed in the study.
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: Tuberculosis is an aerosol transmitted disease of human beings caused by Mycobacterium tuberculosis (Mtb). The only available vaccine for Mtb is Bacillus Calmette-Guérin (BCG). Currently no alternative or booster is available for BCG. The objective of this predictive approach was based on binding of MHC-I and MHC-II and B cell epitopes of Mtb for mouse host.
Methods: Immunoinformatics approach was used to design subunit vaccine (SV) by joining 8 MHC-I bindings, 6 MHC-II bindings, and 8 B-Cell epitopes with AAV, GPGPG, and KK amino acid linkers, respectively. The efficacy of the SV was enhanced through Mtb protein Rv3763 (LpqH, PDB ID= 4ZJM) as an adjuvant at the N-terminal of SV. The in silico analyses evaluated the SV to predict allergenicity, antigenicity, and physico-chemical properties.
Results: Predictions revealed that SV is non-allergic and highly antigenic. The physico-chemical analysis showed that the SV was stable and basic in nature. The three-dimensional structure of SV was stable with a high binding affinity against the mouse TLR2 receptor. In silico cloning suggested the effective transformation of SV into the eukaryotic expression vector.
Conclusion: This study permits preclinical validation of the designed SV in mouse host to confirm its immunogenic potential and efficacy, which will help in controlling tuberculosis.
Keywords: Immunoinformatics; Docking; Subunit vaccine; Lipoprotein; Tuberculosis
Introduction![]()
Bacille Calmette-Guérin (BCG) is the only available attenuated strain of Mycobacterium bovis against Tuberculosis (TB) [1]. As a vaccine, BCG can prevent neonates and children from the spread of TB [2]. However, the use of BCG vaccine has some severe limitations, as its overall efficacy is still in question [3, 4]. Several controversies about BCG include; failure to provide protection against pulmonary TB in adults, the absence of a booster dose, and difficulty in the differential diagnosis between diseased and vaccinated individuals by standard diagnostic test e.g. tuberculin skin test [5, 6]. Consequently, Mtb has undergone a dormant state in most cases after BCG vaccination and is associated with latent chronic infection development [7]. The latent Mtb becomes active when a person's immune status becomes weak, e.g., individuals with genetic immune defects, aged people, and people taking immunosuppressant drugs [6]. To overcome these issues of the currently available vaccine, the development of an alternative vaccine is essential. It is necessary to design a vaccine that is safer, more immunogenic, and induces long-lasting protection. An answer to this could be a multi-epitope subunit vaccine (SV), which is a substitutive approach to immunization. It identifies the peptide epitopes on immunogens and generates the immunogenic response [6]. For this purpose, synthetic versions of peptide epitopes have been used for engineering the vaccine. Contrary to routine vaccines, SV are non-infective, synthetic, possess zero risks of pathogen activation, and are free from unwanted effects, which are common while using whole organism attenuated vaccine [6].
Mtb lipoproteins are a diverse class of membrane-bound proteins with multiple functions [8]. These proteins have signal peptides which help in their secretion out of a bacterial cell and may cause post-translational modification in the host cell [8]. Immunoinformatics is a newly developed approach that predicts thermodynamically stable linker joined multi-epitopes novel vaccine(s) significantly less cost and time. We have used this approach in our study to predict MHC-I, MHC-II binding, and B cell epitopes to design a SV against tuberculosis. This vaccine was evaluated computationally for its physico-chemical characteristics, structure validation, antigenicity, allergenicity prediction, and in silico cloning in the eukaryotic expression vector. This study will help to provide a new immunogenic vaccine that may have the ability to induce the immune response against tuberculosis if evaluated in animal model, by complementing or boosting BCG challenges.
Methods![]()
Collection of Mycobacterium tuberculosis protein sequences for vaccine design
Primary sequences of 89 lipoproteins of Mycobacterium tuberculosis H37Rv were retrieved from Mycobrowser (database of Mycobacterium spp.) in FASTA format. These protein sequences were employed for scheming multi-epitope SV construct potentiated against TB infection. Experimentally known crystal structure of Mycobacterium tuberculosis Rv3763 (LpqH, PDB ID= 4ZJM) TLR2 agonist protein and its primary sequence were retrieved from Protein Data Bank (PDB) [9], and Mycobrowser database respectively, to be used as an adjuvant.
Prediction of epitopes
MHC-I binding epitopes for all the selected lipoproteins of Mtb were predicted using Immune Epitope Database (IEDB) resources [10]. “IEDB recommended 2.19” prediction method. Six MHC alleles (H-2-Db, H-2-Dd, H-2-Kb, H-2-Kd, H-2-Kk, H-2-Ld) each of 9-mer in length were selected, and peptides were sorted based on their lowest percentile rank. Similarly, the IEDB tool (http://tools.immuneepitope.org/mhcii/) [11] was recruited for predicting MHC-II binding epitopes. Default parameters were used against the mouse (host) with locus H-2-I. All three alleles, namely, H2-IAb, H2-IAd and H2-IEd, were selected for this locus. 15-mer length MHC-II epitopes for mouse alleles were predicted for all the lipoproteins. B cell linear epitopes were predicted using ABCPRED server for the ultimate SV design. ABCPRED (http://crdd.osdd.net/raghava/abcpred/ABC_submission.html) [12] with threshold >0.9 and epitope length of 20 amino acids was used.
Multi-epitope SV construct
Epitopes of lowest percentile rank (high binding affinity), 8 of MHC-I and 6 of MHC-II, were joined together with AAY and GPGPG amino acid linkers, respectively. 8 B cell epitopes with score ≥ 0.95 were selected and joined with KK amino acid linker to form the final vaccine construct. Therefore, mouse TLR2 (Uniprot ID: Q9QUN7) agonist Rv3763/LpqH Mtb protein [9] was selected as an adjuvant to augment the antigen-specific immune reactions. Moreover, a 13 amino acid sequence (AKFVAAWTLKAAA) known as PADRE sequence was concatenated at the start and the end of the epitope sequence of the vaccine to enhance its efficacy [13, 14]. The final SV consisted of 593 amino acids.
Physiochemical parameter, allergenicity, and antigenicity predictions of SV construct
Physicochemical parameters of the vaccine were determined by using ProtParam [15] web server, a part of Expert Protein Analysis System (EXPASY). Various parameters were predicted using this tool, including; theoretical pI, estimated half-life, molecular weight (kDa), grand average of hydropathy (GRAVY), and aliphatic index. AlgPred web tool [12] was applied for the prediction of allergenicity of SV. AlgPred uses combined algorithms (SVMc+ IgEepitope + ARPs BLAST+ MAST) approach to give highly accurate results. Allergenicity measurement of a protein is essential because most of the allergens are protein in nature and can cause sensitization or severe allergic reactions. Thus, the predicted vaccine construct must be free from the allergenic nature. For antigenicity index prediction, ANTIGENpro tool was applied, as this tool is efficient and accurate. ANTIGENpro performs prediction by using primary protein sequences regardless of any pathogen and without performing their alignment. For categorizing a protein as antigenic or non-antigenic, ANTIGENpro uses a support vector machine (SVM) algorithm based on its microarray data.
Prediction of SV secondary structure
For secondary structure prediction of SV protein, PSIPRED web-based server 4.0 (http://bioinf.cs.ucl.ac.uk/psipred/) was applied, which uses Psi-BLAST i.e., Position-specific iterated BLAST method to find out the closest identical sequences of related protein templates or sequences.
Tertiary structure prediction of SV construct and molecular docking with TLR2 immune receptor
Tertiary structure prediction of the vaccine is crucial because it is more stable, and this stability is achieved by decreasing the energy of protein molecules. Three-dimensional SV vaccine structure was designed and configured by RaptorX server at default parameters. Energy minimization and structure visualization were carried out by (University of California, San Francisco) UCSF Chimera tool. To perform the interaction analysis, a molecular docking approach was used in which SV construct was docked against mouse TLR2 (PDB ID: 2Z7X) using PatchDock (http://bioinfo3d.cs.tau.ac.il/PatchDock/) [16] followed by the rescoring and refinement of docked solutions through FireDock (Fast Interaction Refinement in Molecular Docking) server. FireDock predicts the ten best way-outs based on binding score. Interacting amino acids of SV and TLR2 receptors were visualized through the LigPlot tool.
It is crucial to detect whether a designed vaccine has a binding affinity with its receptor or not [17]. If the receptor can recognize Mtb, then it leads to an immunogenic response in the host [18].
Reverse translation, codon optimization and in silico cloning of SV construct
To check the expression of the designed SV construct in the eukaryotic system, we first reverse-translate our protein using Sequence Manipulation Suite: reverse translation tool, and the resulting sequence was used for codon optimization. Java Codon Adaptation Tool (JCAT) [19] was used to optimize the codon of SV construct in eukaryotic organisms (Mus musculus). Three additional parameters, including prokaryote ribosome binding site, restriction enzymes cleavage sites, and avoid the rho-independent transcription termination, were selected for appropriate codon optimization, followed by XhoI restriction site digestion and cloning into adeno-associated virus vector (pAAV) by using SnapGene tool.
Results![]()
Epitopes collection and design of SV
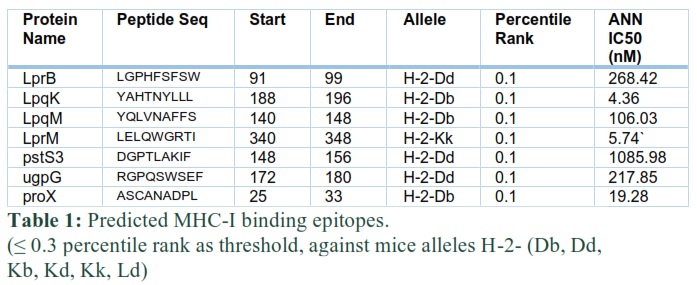
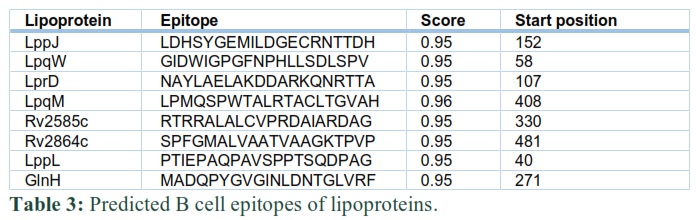
After predicting MHC-I binding, MHC-II binding, and B cell epitopes, the immuno-informatics approach was used to construct a SV. MHC epitopes were selected according to the instructions of IEDB i.e. low percentile epitopes have a high binding affinity with MHC molecules. Thus, for percentile rank, a threshold of less than or equal to 0.3 was self-created for shortlisting of epitopes of different proteins. The shortlisted MHC-I and MHC-II binding epitopes for lipoproteins are presented in table 1 and 2, respectively. B cell epitopes were selected based on their highest score. To shortlist these epitopes, a self-created threshold of more than 0.9 score was used in table 3.
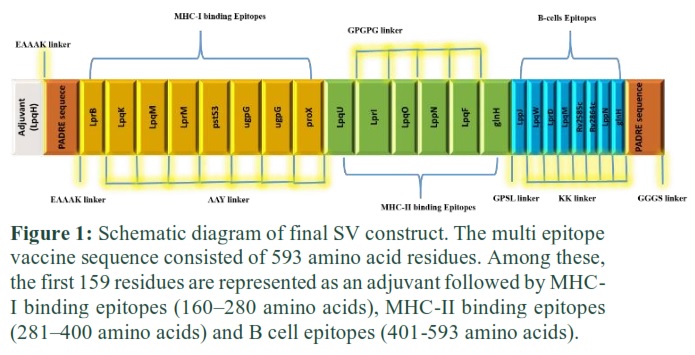
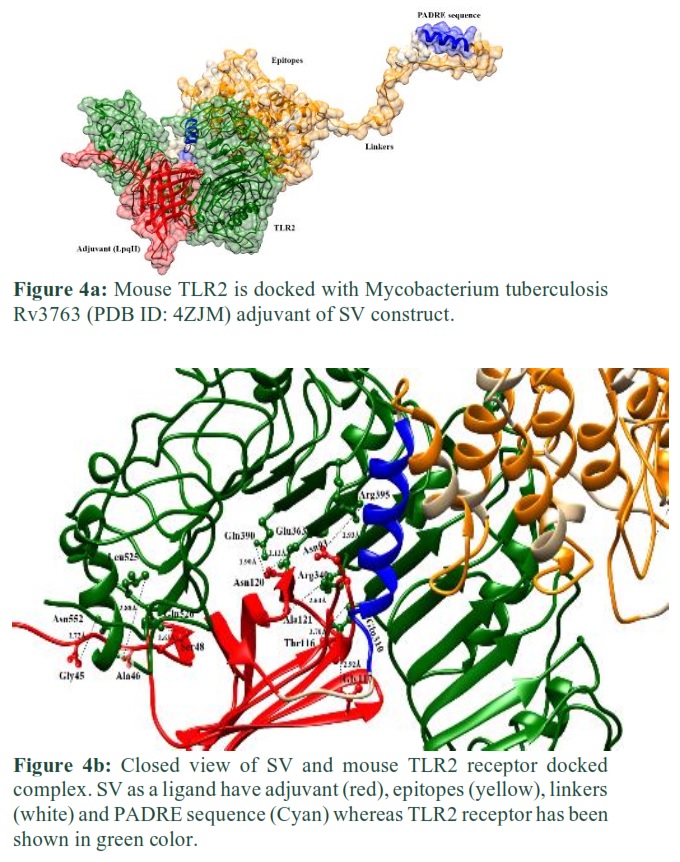
These epitopes were linked together by using a linker in such a way that MHC-I epitopes are linked by AAY, MHC-II by GPGPG, and B cell epitopes by KK linkers, respectively. Further, the Rv3763 (LpqH, PDB ID= 4ZJM) protein of Mtb [9] was used as an adjuvant at N-terminal to improve the immunogenicity of the vaccine. A sequence of 13 amino acids, the PADRE sequence [14], was incorporated after adjuvant and at the end of the last epitope to further enhance vaccine efficacy (Fig. 1).
This Diagram clearly showed that the EAAAK linker joined the adjuvant and MHC-I binding epitopes, while MHC-I and MHC-II binding epitopes were connected through AAY and GPGPG linkers. The MHC-II binding epitope and first B cell epitope were joined by a GPSL linker.
Allergenicity, Antigenicity and Physicochemical Predictions of vaccine
The results of allergenicity prediction by AlgPred revealed that SV is non-allergen with a -0.69 score, which is less than the threshold value of -0.4. However, antigenicity prediction by ANTIGEN pro revealed that SV is a probable antigen with a score of 0.7730, which is more than the threshold value of 0.4. Physico-chemical properties were obtained by using ProtParam, which showed that SV protein has a total of 593 amino acids with a molecular weight of 60.1 kDa. The theoretical PI is 9.89, which depicts that our SV is basic in nature, while the computed instability index of SV is 37.31, demonstrating that SV is stable in nature. The grand average of GRAVY value is -0.018 which means that SV is hydrophilic i.e. easily dissolvable in water.
Secondary Structure Prediction
The secondary structure of SV protein was predicted by PSIPRED server (Fig. 2). The results were obtained in the form of a strand (yellow), helix (pink), and coil (grey line), which are presented with different color schemes. Percentage of strand, helix and coil in SV is 17.87%, 40.64%, and 41.49%, respectively.
Prediction and validation of the tertiary structure in protein
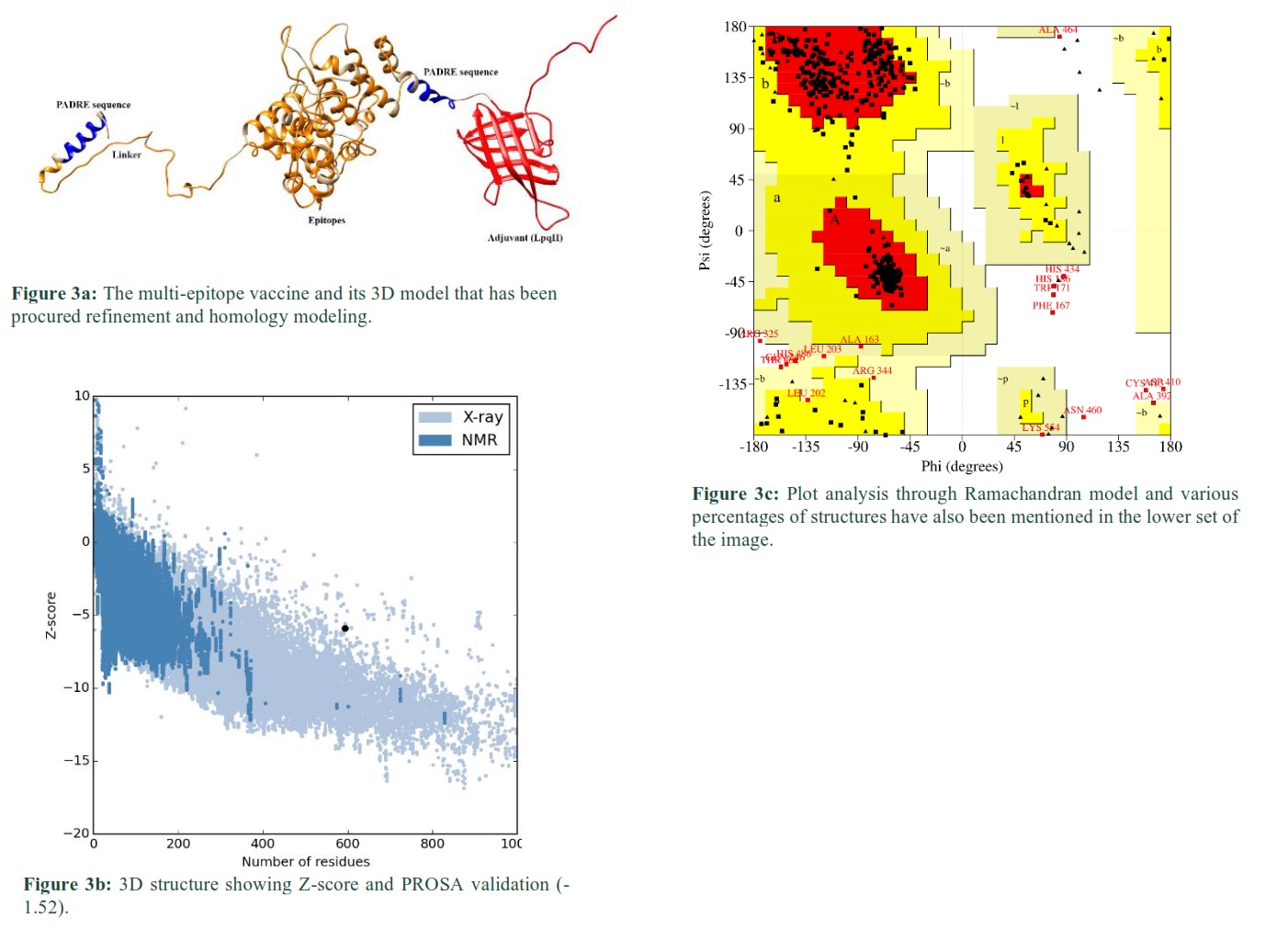
This pattern of the protein was predicted through RaptorX and the resultant SV has been shown in Fig. 3a. 3D model of SV was predicted for 3 domains at different amino acid, including; domain 1 (1-268) 2.40 x 10-10, domain 2 (366, 581) 8.60 x 10-5and domain 3 (269, 365) 1.20 x 10-4. The calculated overall un-normalized GDT (uGDT) of SV structure was 347. 100% residues (593) in the SV protein were modeled and 11% (64) residues were in the disordered region.
The quality or checking of any error in the 3D model of protein was performed using ProSA-web [26]. ProSA-web calculated Z-score -1.52 for SV (Fig. 3b). The Ramachandran plot validated the tertiary structure of SV. The plot showed (Fig. 3c) that 423 out of 593 (87.9%) residues were in the favored regions [A, B, L], 40 residues (8.3%) were in additional allowed regions [a,b,l,p], 7 (1.5%) residues were in generously allowed regions [~a, ~b, ~l, ~p] and only 11 (2.3%) residues were in disallowed regions. The number of glycine residues (shown in a triangle) was 69, and the number of proline was 41. Thus, this tertiary structure is of good quality as most of its residues are present in the most favored regions.
Docking of SV with TLR2 receptor
Molecular docking was performed through PatchDock server. SV protein was docked with mouse TLR2 receptor containing adjuvant. PatchDock server produced docked solutions based on refined geometry and electrostatic affinity of the protein surface. Resulting docked complexes were subjected to Fast Interaction Refinement in Molecular Docking (FireDock) server for refinement and re-scoring based on respective binding energy. The top-ranked complex was selected, having global energy -7.46 and attractive Van Der Waals interactions score -11.71 (Fig. 4a).
U-shaped binding pocket amino acid residues (Glu310, Arg340, Arg395, Glu363, Gln390, Leu 525, Gln526 and Asn552) of TLR2 receptor were involved in hydrogen bonding with SV residues (Gly45, Ala46, Ser48, Asn93, Thr116, Gly117, Asn120, and Ala121) after molecular docking. The docked result is shown in Fig. 4b.
Reverse translation and codon optimization
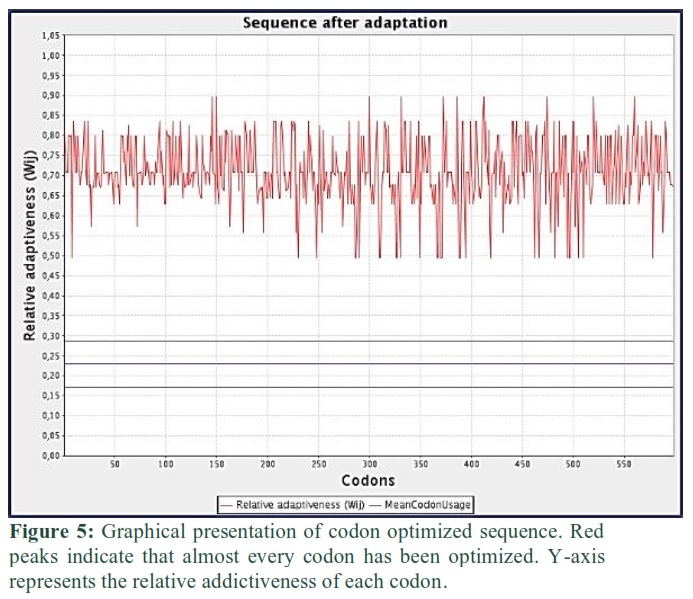
SV sequence was reverse translated to the nucleotide sequence. The total length of the reverse translated sequence is 1794 bp. Then this sequence was codon-optimized for expression in the mice host. Codon adaptation index (CAI) value of optimized sequence was 0.71, GC content of improved sequence was 73.24 and GC content of Mus musculus was 41.76%. Graphical representation of optimized or adapted sequence is shown in Fig. 5.
In silico optimization and cloning of vaccinal protein
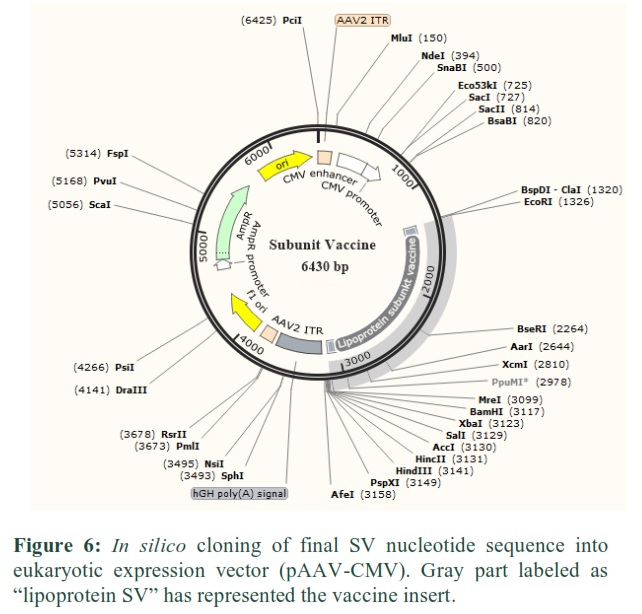
The SV nucleotide sequence was codon adapted/optimized according to the mammalian (mouse) vector by introducing BsmI and BamHI restriction sites in N- and C-terminal of the vaccine sequence. Finally, the improved sequence with restriction sites was adjusted in the pAAV-CMV-eGFP vector by means of SnapGene tool to guarantee protein expression in the vaccine (Fig. 6).
Figures & Tables


Discussion![]()
Vaccination is an efficient way to control infectious diseases and improve public health [20]. Now-a-days, much genomic, transcriptomic, and proteomic information is available for Mtb and other pathogenic microbes. Computational approaches were applied using this information to identify potential antigenic targets [21]. In our study, we focused to design a SV from 89 lipoproteins of Mtb. Instead of using the whole organism as a vaccine agent, only the immunogenic part of these proteins was used to construct the vaccine. Lipoproteins of Mycobacterium tuberculosis belong to the functionally broad class of membrane-bound proteins [8]. These proteins are involved in providing virulence and immunogenicity to Mtb as the interruption in the biosynthesis of lipoproteins leads to the weakening of Mtb activity [22].
Mtb epitopes that showed resemblance with the host proteome were identified. The protein sequences of lipoproteins were imperiled to BLASTp search against the human host proteome. These proteins of Mtb did not show any sequence homology with human proteins. Thus, these proteins could be excellent immunogenic targets for synthesizing vaccines against Mtb. In this study, epitopes for B and T cells of 89 lipoproteins were predicted. Epitopes of T cells are presented on the surface of antigen-presenting cells by combining with MHC molecules [23]. MHC molecules are antigen-presenting moieties, mainly of two types, i.e. class I and class II [24]. Class I molecules, mainly represented to cytotoxic T lymphocytes or CD8+ T cells, can be presented on any kind of nucleate cell that represents the endogenous processed protein or the engulfed processed protein that were digested and processed in cytosolic pathways [25]. Class II molecules are mainly combined and present exogenous proteins, such as pathogens’ surface proteins. These are processed in the cellular endocytic pathways and presented to helper CD4+ T lymphocytes [20].
The immunoinformatics approach to analyze SV demonstrated that our final SV protein has a tremendous binding affinity with MHC-I, MHC-II, and B-cell epitopes based on structural and physicochemical characteristics. Model quality of the 3-dimensional vaccine protein structure predicted by homology modeling suggested the refined amino acid distribution. Ramachandran plot shows that most of the vaccine residues are present in the favorable region, and very few are located in the disallowed regions. This showed that the overall model quality of protein structure is acceptable.
The conformational stability of the vaccine was done by docking with the TLR2 receptor to determine the immunological response of the vaccine construct. Energy consumption was reduced to decrease the potential energy of the protein-receptor complex system. This process also keeps the dismissed arrangements of protein structure by replacing a few subcellular structures in the protein to yield appropriate stereochemistry of a stable protein framework.
The optimum level of translation expression of a vaccine in the eukaryotic system is necessary for vaccine functionality
Tuberculosis disease has become an intense problem at the international level. It causes many deaths in the developing states of Asia and Africa, and there is still no permanent control and remedial measure for the disease. The immunoinformatics technique was used for the development of a multi-epitope SV. This work begins with the recovery protein sequences of 89 Mtb lipoproteins. After that the immunogenic B- and T-cell epitopes were predicted, epitopes were joined by linkers for the effective separation of epitopes in in-vivo system, adjuvant was added in the start to enhance the immunogenicity and then design the SV to get the cell-mediated and humoral immunity. Allergenicity and antigenicity parameters were predicted to confirm that the vaccine is non-reactive in nature. The binding affinity with the host cell receptor was evaluated by molecular docking. Finally, the effective expression of SV protein was made sure by in silico cloning. Furthermore, the recommendation obtained from this study declares that the proposed vaccine requires experimental validation to analyze the efficacy of the vaccine in the sense of generating adequate immunological memory.
Author Contributions
MMA and MAN conceived and designed this study; MAN, MMA and AK performed the experiments; SA, QA, WA, AS and WA analyze the data/results; MAN, MMA and SA wrote the manuscript. All authors read and approved the final manuscript.
None.
Availability of data and materials
Please contact author for data requests.
Acknowledgements
Authors would like to thank Huazhong Agricultural University, Wuhan, China for providing facilities for this study. Authors are also thankful to Sonakshee Havis, New York University School of Global Public Health USA for improving the manuscript in terms of English grammar and language.
References![]()
- World Health Organization. BCG vaccine: WHO position paper, February 2018-recommendations. Vaccine, (2018); 36(24): 3408-3410.
- Gideon HP, Flynn JL. Latent tuberculosis: what the host “sees”? Immunologic research, (2011); 50(2-3): 202-212.
- Andersen P, Doherty TM. The success and failure of BCG—implications for a novel tuberculosis vaccine. Nature Reviews Microbiology, (2005); 3 (8): 656-662.
- Fine PE. Variation in protection by BCG: implications of and for heterologous immunity. The Lancet, (1995); 346 (8986): 1339-1345.
- Crampin AC, Glynn JR, Fine PE. What has Karonga taught us? Tuberculosis studied over three decades [State of the art series. Tuberculosis. Edited by ID Rusen. Number 4 in the series]. The international journal of tuberculosis and lung disease, (2009); 13(2): 153-164.
- Glick BR, Delovitch TL, Patten CL. Chapter 11: Vaccines: Medical Biotechnology. 2014; 632-663. American Society Microbiology Press. Washington DC.
- Flores-Valdez Mario Alberto. Why is it important to improve vaccines against latent tuberculosis? Frontier for young mind. https://kids.frontiersin.org/article/10.3389/frym.2016.00019 (2016).
- Sutcliffe IC, Harrington DJ. Lipoproteins of Mycobacterium tuberculosis: an abundant and functionally diverse class of cell envelope components. FEMS microbiology reviews, (2004); 28(5): 645-659.
- Noss EH, Pai RK, Sellati TJ, Radolf JD, Belisle J, Golenbock DT, Boom WH, Harding CV. Toll-like receptor 2-dependent inhibition of macrophage class II MHC expression and antigen processing by 19-kDa lipoprotein of Mycobacterium tuberculosis. The Journal of immunology, (2001); 167(2): 910-918.
- Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics, (2016); 32 (4): 511-517.
- Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, Peters B. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC bioinformatics, (2010); 11(1): 568.
- Saha S, Raghava GP. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic acids research, (2006); 34(suppl_2): 202-209.
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic acids research, (2007); 35(suppl_2): 407-410.
- Wierecky J, Müller MR, Wirths S, Halder-Oehler E, Dörfel D, Schmidt S., Häntschel M, Brugger W, Schröder S, Horger MS, Kanz L. Immunologic and clinical responses after vaccinations with peptide-pulsed dendritic cells in metastatic renal cancer patients. Cancer research, (2006); 66(11): 5910-5918.
- Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: The proteomics protocols handbook. 2005; 571-607. Humana press.
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic acids research, (2005); 33(suppl_2): 363-367.
- Pandey RK, Kumbhar BV, Srivastava S, Malik R, Sundar S, Kunwar A, Prajapati VK. Febrifugine analogues as Leishmania donovani trypanothione reductase inhibitors: binding energy analysis assisted by molecular docking, ADMET and molecular dynamics simulation. Journal of Biomolecular Structure and Dynamics, (2017); 35(1): 141-158.
- Drage MG, Pecora ND, Hise AG, Febbraio M, Silverstein RL, Golenbock DT, Boom WH, Harding CV. TLR2 and its co-receptors determine responses of macrophages and dendritic cells to lipoproteins of Mycobacterium tuberculosis. Cellular immunology, (2009); 258 (1): 29-37
- Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn D. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic acids research, (2005); 33 (suppl_2): 526-531.
- Skwarczynski M, Toth I. Peptide-based synthetic vaccines. Chem Sci [Internet] (2016); 7(2): 842-854.
- Purcell AW, Mccluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nature Revews Drug Discovery, (2007); 6(5): 404-414.
- Becker K, Sander P. Mycobacterium tuberculosis lipoproteins in virulence and immunity–fighting with a double‐edged sword. FEBS letters, (2016); 590(21): 3800-3819.
- Rothbard JB, Taylor WR. A sequence pattern common to T cell epitopes. The EMBO journal, (1998); 7(1): 93-100.
- Janeway C, Travers P, Walport M, Shlomchik M. Chapter 5: Antigen presentation to T lymphocytes. Immunobiology, (2001); 5: 168.
- Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nature Reviews Immunology, (2012); 12 (8): 557-569.
- Makrides SC. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiology and Molecular Biology Reviews, (1996); 60(3): 512-538.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0

