

Full Length Research Article
Structural interpretation of the NS3 helicase ATP binding domain of Zika virus (ZIKV)
Afnan Mohammed Shakoori1*, Nida Alsaffar2
Adv. life sci., vol. 11, no. 1, pp. 144-152, February 2024
*– Corresponding Author: Afnan Mohammed Shakoori (amshakoori@uqu.edu.sa)
Authors' Affiliations
2. Department of Medical Laboratory Sciences, Mohammed Al-Mana College of Medical Sciences, Al Safa, Dammam – Saudi Arabia
[Date Received: 25/08/2023; Date Revised: 16/01/2024; Date Published: 25/02/2024]
Editorial Note on Version of Record
31 May 2025: This article has been corrected. See https://doi.org/10.62940/als.v13i0.4283 for more information.
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: The Zika virus, a mosquito-borne virus, was discovered in Uganda and quickly spread to Asia and the Pacific. Zika is mainly transmitted by the bite of an infected Aedes species mosquito (Aedes aegypti and Aedes albopictus). These mosquitos bite both during the day and at night.
Methods: Numerous tools (SMART, Pfam, InterProScan and Scan Prosite, BepiPred-2.0 server, I-TASSER, PROCHECK, phyre 2, protparam, and GPS 6.0) were used to elaborate the content in this publication.
Results: The results show that HABD (187 amino acids) is a DEAH-Box RNA helicase. We also used the Ramachandran plot to validate the epitope peptides and structure modeling. We also discovered the amino acid composition and various residues/parentages in the HABD protein phosphorylation site prediction and PK-specific phosphorylation sites (p-sites).
Conclusion: The helicase ATP binding domain (HABD), HABD protein ATP binding sites, and epitope binding peptides are discussed in this work. There are five atoms in total: nitrogen, sulfur, hydrogen, carbon, and oxygen. Hydrogen atoms (1476) provide the most to the composition. We created a graph representing the protein’s predicted phosphorylation sites (p-sites). Aside from the standard statistics, GPS 6.0 may identify PK-specific p-sites hierarchically. GPS 6.0 could be a valuable service for further phosphorylation study.
Keywords: DEAH-Box; Epitope peptides; Ramachandran plot; Structure Modelling; Secondary structure; Phosphorylation; PK-specific and GPS 6.0
Introduction![]()
Zika virus (ZIKV) is a member of the Flaviviridae family and the Flavivirus genus. With 53 species, the flavivirus genus is the largest in the Flaviviridae family and can be further subdivided into non-vector, tick-borne, and mosquito-borne clusters [1,2]. Mosquito-borne flaviviruses such as ZIKV, dengue virus (DENV), West Nile virus (WNV), yellow fever virus (YFV), and Japanese encephalitis virus (JEV) have been a growing public health concern in the recent decade due to an increase in global incidence [1,2]. ZIKV traveled across the Pacific Ocean in May 2015 and was introduced in Brazil, where it caused over one million cases [3]. As of May 2019, the virus had spread swiftly to 84 nations, territories, or subnational areas, posing a global public health threat [4]. ZIKV’s genome is a 10.8-kb single-stranded positive-sense RNA molecule with a 100-nucleotide 5′ untranslated region (UTR), a single 10 kb open reading frame (ORF), and around 420 nucleotides in the 3′ UTR. The ORF encodes a significant polyprotein precursor of 3423 amino acids, which is then co- and post-translationally cleaved into three structural (C, prM, and E) and eight nonstructural (NS1, NS2A, NS2B, NS3, S4A, NS4B, & NS5) proteins [5]. The N-terminal protease and C-terminal helicase domains of NS3 are structurally distinct functional domains [6]. Serine protease, NTPase, and RNA helicase (ATPase and Unwinding activity) are all found in NS3.
Antigen-antibody interaction is critical in immunological processes and reactions within the immune system, & B-cell epitopes are antigen regions recognized and bound by B-cell-produced antibodies [7]. Additionally, in the presence of particular cross-reacting antibodies, B-cell epitopes can stimulate the humoral immune response [8,9]. The method of quickly identifying immunological epitopes is based on computational prediction, which employs advanced computations and an increasing epitope information base. The phosphorylation potential of ZIKV proteins has yet to be determined. Understanding the phosphorylation potential of viral proteins is critical since it aids in viral replication and interaction with host receptors [10]. This study provides molecular insight for future research into the structural and functional characteristics of the HABD protein. Protein phosphorylation is required for several biological functions. Protein phosphorylation is one of the most researched posttranslational modifications (PTMs) that control physical processes [11]. Protein kinases catalyze the reversible covalent attachment of a phosphate group on particular serine, threonine, and tyrosine residues of proteins [12, 13, 14]. Many in silico prediction approaches, in addition to experimental screening, have been developed to discover PK-specific phosphorylation sites (p-sites) efficiently and efficiently. From 2004 to 2017, we created various GPS algorithms for p-sites and other PTM site predictions, ranging from version 1.0 to 4.0 [15, 16, 17, 18]. 2020, our team will produce GPS 5.0, combining position weight determination and score matrix optimization modules. GPS 5.0 could predict the p-sites of 479 human PKs inside the classical module [19]. Identifying potential phosphorylation sites and the protein kinases that bind to them is critical for understanding how many proteins work. This paper describes GPS 6.0, a prediction system that predicts phosphorylation sites in a kinase-family-specific manner. We can now explore phosphorylation-dependent processes and probe the kinases involved in controlling these functions since we have a predictor that effectively incorporates the context and sequence conditions that govern phosphorylation.
Methods![]()
The arrangement of the helicase ATP binding domain (HABD)
The 187 amino acid sequence of the Zika Virus Helicase ATP binding domain protein detailed domain arrangement. One of the critical domains is the crucial (HABD) helicase ATP binding domain. For this study, the sequence of this HABD domain was retrieved from the NCBI genome database. We use SMART, Pfam, InterProScan, and Scan Prosite the most for manual domain categorization [20,21,22]. We also used the BepiPred-2.0 server to predict B-cell epitope peptides from the sequence [23].
I-TASSER is used to predict 3-D structure
The amino acids of the 187 Helicase ATP binding domain (HABD) sequence were obtained from a database in FASTA format. The 3-D (Three-dimensional) model was created with the I-TASSER accessible service, which builds a three-dimensional model of the query sequence using multiple threading alignments and iterative structure assembly simulation [24]. The details for choosing this server were its accessibility, combined modeling technique, & success in the CASP competition. Threading, structural complex, model selection, refining, & structure-based efficient annotation are all general processes in the I-TASSER technique [25]. First, the evolutionary related protein was tested against the query sequence using PSI-BLAST, which generated a sequence profile that PSIPRED used to estimate secondary structure [26]. The request sequence was then LOMETS [27] routed by the illustrative PDB structure collection. Z-score was used to assess the excellence of template alignment, and the best one was chosen for further examination. The constant pieces in threading alignments were removed from an assembled structural model of aligned regions in the following phase [24]. Unaligned region modeling accuracy is generally low, whereas threading aligned area modeling accuracy is high. Thus, these template fragments remain rigid throughout the simulation to get a high-resolution structure. For fragment assembly, the replica exchange Monte Carlo simulation technique was used [28]. C/side chain correlation, H-bonds, hydrophobicity, spatial restraints from threading templates [28], & sequence-based contact predictions from SVMSEQ [29] are all included in the refinement simulation-generated conformations [30]. The designated cluster centroids were employed over in the refinement phase to do additional fragment association simulation, which helps to reduce steric conflicts and refine the global topology of the cluster centroids. TM-align [31] was used to identify the PDB structures structurally near the cluster centroids. REMO [32] was used to create the final structural models, which employed cluster centroids from the second simulation round as input. In the final phase, the function of the three-dimensional model of the request protein was anticipated by comparing it to proteins with known structure & function in the PDB. The functional analogs were ordered according to their TM score, RMSD, sequence identity, and structural alignment coverage. The C-score (confidence score) measured the anticipated model’s quality, which varied from 5 to 2. It is determined by threading alignment quality & the convergence of structural association refinement simulations.
Validation of structure models
The Ramachandran plot confirmed the conformation of the finest Helicase ATP binding domain (HABD) model anticipated by I-TASSER tool. The expected model’s conformation was derived by means of examining the phi () and psi () torsion angles with the PROCHECK web service. PROCHECK’s Ramachandran plot designates a high-grade model with over 90 percent residues in the maximum favored region [33].
Prediction of Secondary structure
The Helicase ATP binding domain (HABD) (187 amino acids) structure was demolished exhausting the phyre 2 server (http://www.sbg.bio.ic.ac.uk/phyre2). Phyre2 is a web-based tool for expecting & analyzing protein structure, function, & mutations [34]. A representative structural expectation will be returned between 30 minutes & 2 hours of submittal. The availability of beta strands, -helix, and disordered in the HABD region is substantial [20, 21].
Analysis of the HABD protein sequence and its content
A Zika virus helicase ATP binding domain amino acid sequence (1-187) was submitted to ExPASy Server via the link (https://web.expasy.org/protparam/), and data was produced [35,36]. The whole amino acid content of the protein (in CSV format) is entered into an excel spreadsheet, and a graph is drawn to indicate the atomic design, which means the number of atoms in this protein (HABD). Similarly, we conducted extensive research to find positively and negatively charged amino acids. The sequence of helicase ATP binding protein was analyzed using the accessible link (NsitePred webserver. or http://biomine.cs.vcu.edu/servers/biomine.php?id=20230714153327) for ligand prediction and critical probability with the score. The server is intended for predicting sequence-based binding residues for ATP, ADP, AMP, GDP, and GTP [37].
The kinase-specific phosphorylation site prediction
Phosphorylation is the best important post-translational modification in eukaryotes, & it shows an essential role in various cellular functions. A computer prediction method is gaining popularity as a basic complementary strategy in researching protein phosphorylation sites. Prediction tools are classified into two types: Tools for kinase and non-kinase analysis (http://sysbio.unl.edu/PhosphoSVM). A kinase-specific expectation tool takes a protein sequence & the kinase type as input & outputs some measure of the probability that each S/T/Y residue in the sequence is phosphorylated via the chosen kinase [38- 41].
GPS 6.0 predicts protein/peptide phosphorylation
Protein phosphorylation, performed via protein kinases (PKs), is one of the essential PTMs and governs practically all biological activities. We used the FastA format of the HABD protein sequence to submit to the tool (https://gps.biocuckoo.cn/online.php) for p-site prediction. Then, choose the kinase node(s). The predictor has 10 S/T groups, 1 Y group, and 2 Dual groups for dual-specificity kinases. Press the “Submit” button and wait a bit. The extrapolation results of kinase-specific phosphorylation sites on the substrate are available. Protein statistics and disorder tendencies are also provided [42-44].
Results![]()
The Zika Virus Genome Organization and its HABD Critical Domain
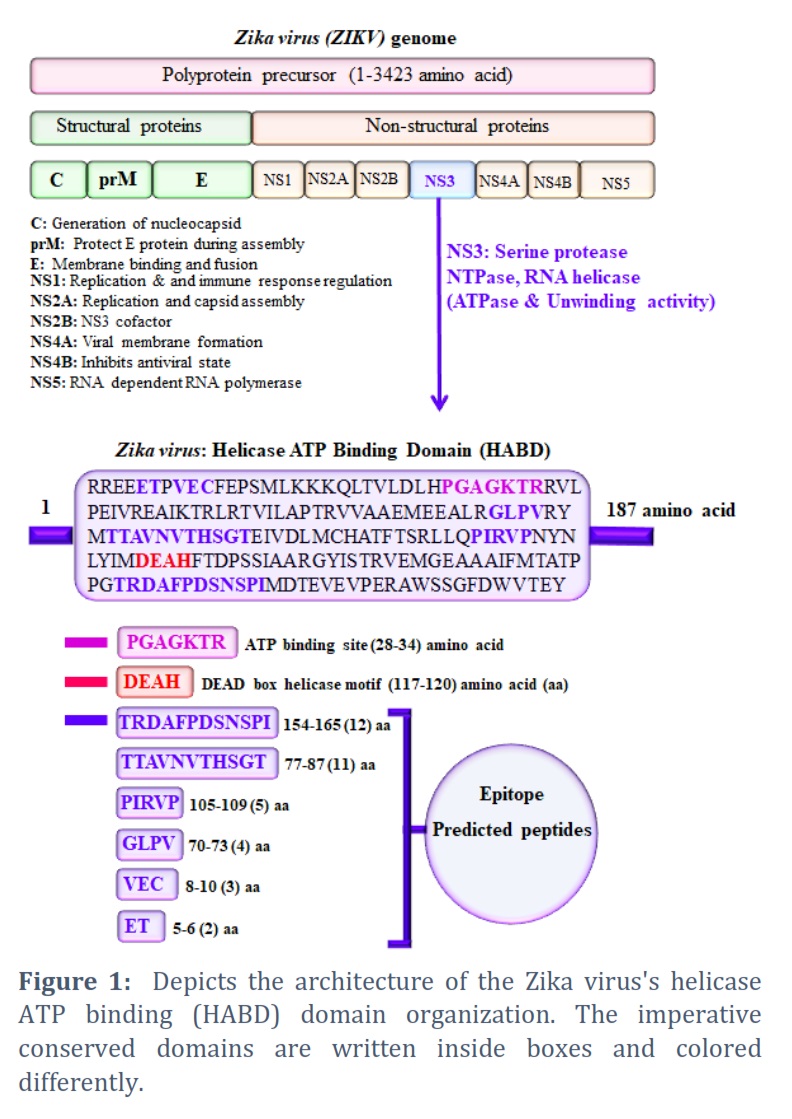
The genome of ZIKV (Zika virus) is a single-stranded (ss) positive-sense RNA molecule that comprises approximately 3423 amino acids of a polyprotein precursor [45]. It is hewn into three structural proteins (C, prM, and E) & eight nonstructural proteins (NS1, NS2A, NS2B, NS3, S4A, NS4B, & NS5) (Figure 1). The role of each domain (structural and nonstructural protein) is indicated in the domain architect below (left side). The NS3 domain (Helicase domain) and its binding sites were the focus of this manuscript. The Helicase ATP binding domain (HABD) organizations were discovered utilizing available bioinformatics tools/servers. We found the HABD with 187 amino acid stretches (Figure 1). The HABD belongs to the DEAH-Box RNA helicase family. ATP binding site length (28-34 amino acids), DEAD box helicase motif length (117-120 amino acids). Similarly, we found the epitope peptides in the helicase ATP binding domain, depicted in blue and emphasized with blue color boxes (Figure 1).
HABD protein 3-D structural analysis
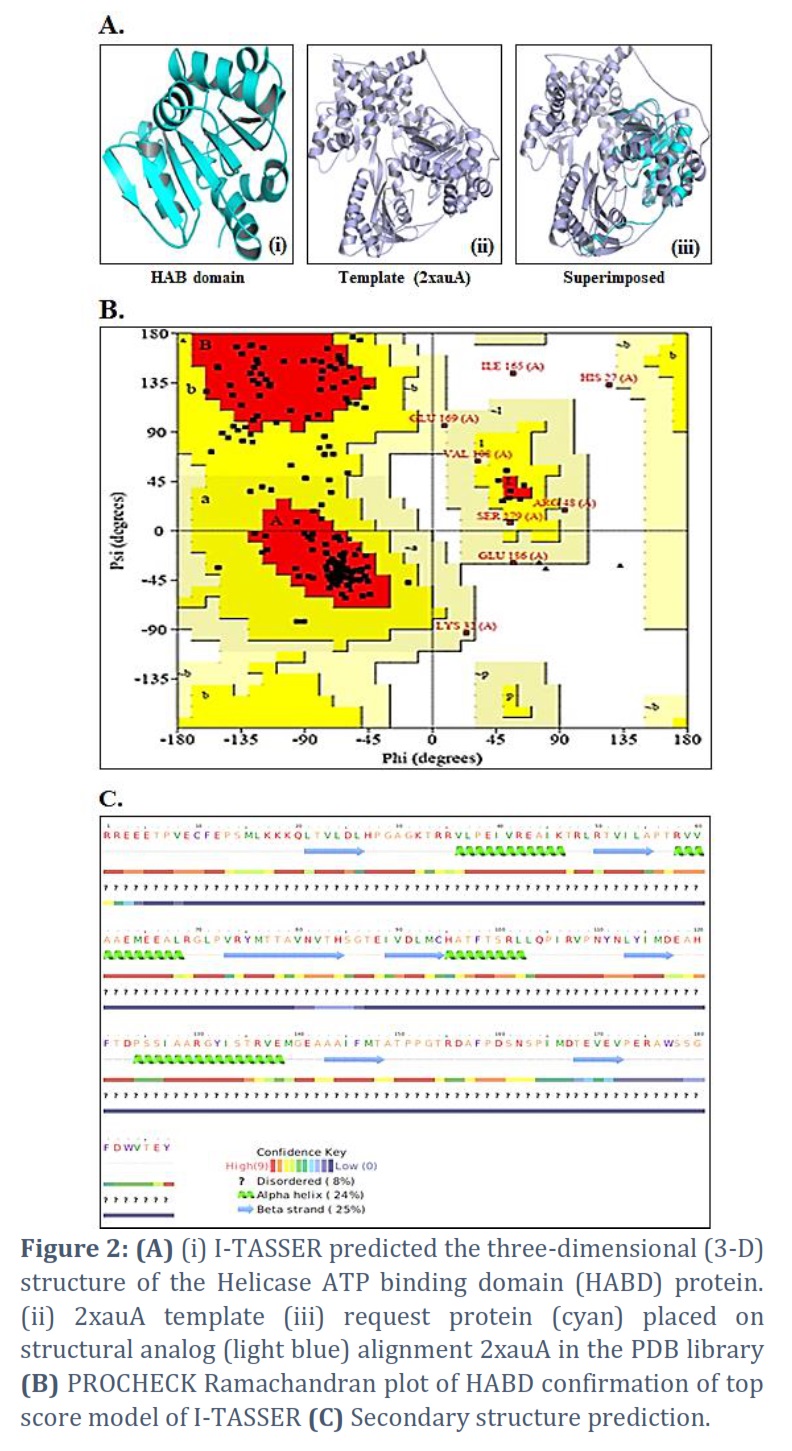
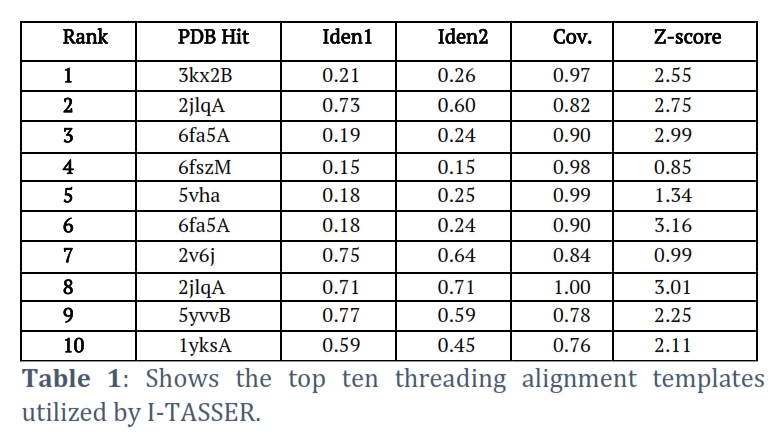
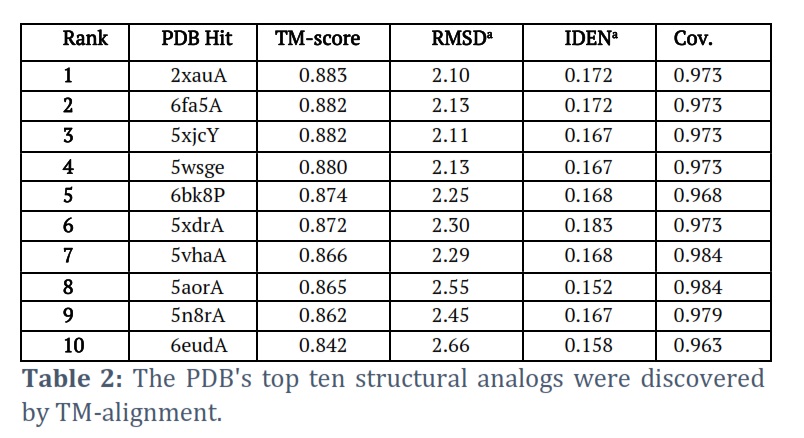
I-TASSER successfully got the projected model of the Helicase ATP binding domain (HABD) protein & its 3-D coordinate file in PDB format. The server returned an anticipated secondary structure using a confidence score (ranging from 0 to 9), indicated solvent approachability, five anticipated facilities by way of C-score, top ten PDB templates within alignment, top ten PDB structural, functional analogs protein, & binding site residues. Model HABD (Figure 2 A) was chosen as the finest C-score and RMSD model. C-scores with higher values indicate a higher-quality model. The top ten threading templates for request protein sequence HABD were recognized by way of the LOMETS meta-server (Table 1). The threading alignment is commonly expected by normalized Z-score. However, a standardized Z-score >1 value reflects a confident alignment. Still, in the case of minor alignment of the extensive request sequence, it does not give a momentous sign of modeling accuracy. Then, the percentage sequence identity in the threading aligned region (Iden1) & the whole chain (Iden2) was careful about excellent homology (Table 1). With a TM-score of 0.883, the structural alignment program TM-align found that 2xauA in the PDB collection was the best structural analog of the highest-scoring model of I-TASSER (Table 2).
The sequence identity percentage of the templates in the threading aligned region with the query sequence is called Ident1. The percentage sequence identity of all the template chains with the query sequence is called Ident2. The threading alignment coverage is calculated by dividing the total number of aligned residues by the query protein’s length. The threading alignments’ normalized Z-score is known as the Z-score. A good alignment is indicated by an alignment with a Normalized Z-score >1 and vice versa.
A scale for comparing two structures’ structural similarity that was recently proposed is called the TM-score. The RMSD between residues that TM-align has structurally aligned is known as RMSDa. In the structurally aligned region, IDENa stands for the percentage sequence identity. Cov, the number of structurally aligned residues divided by the query protein’s length, indicates the alignment’s coverage by TM-align.
HABD protein predicted model evaluation
PROCHECK servers provided Ramachandran plots of the finest anticipated model, demonstrating the model’s dependability. The PROCHECK Ramachandran plot revealed 95.1% residues in the most favored regions & 3.7% in further authorized areas, for a total of 98.8% residues in allowed regions (Figure 2 B).
HABD protein secondary structure prediction
Techniques for predicting secondary structures were evaluated. The Helicase ATP Binding Domain (HABD) has strand, -helix, and disordered secondary systems, according to the overall secondary structure investigation (Figure 2C). Elemental data, for example, secondary or tertiary structure predictions, can be used to characterize an underlying theory. Data on folds collected by biophysical techniques (e.g., circular dichroism) or functional data (existence of a conserved motif/domain) can also inform an initial strategy.
Analysis of HABD protein sequences and composition
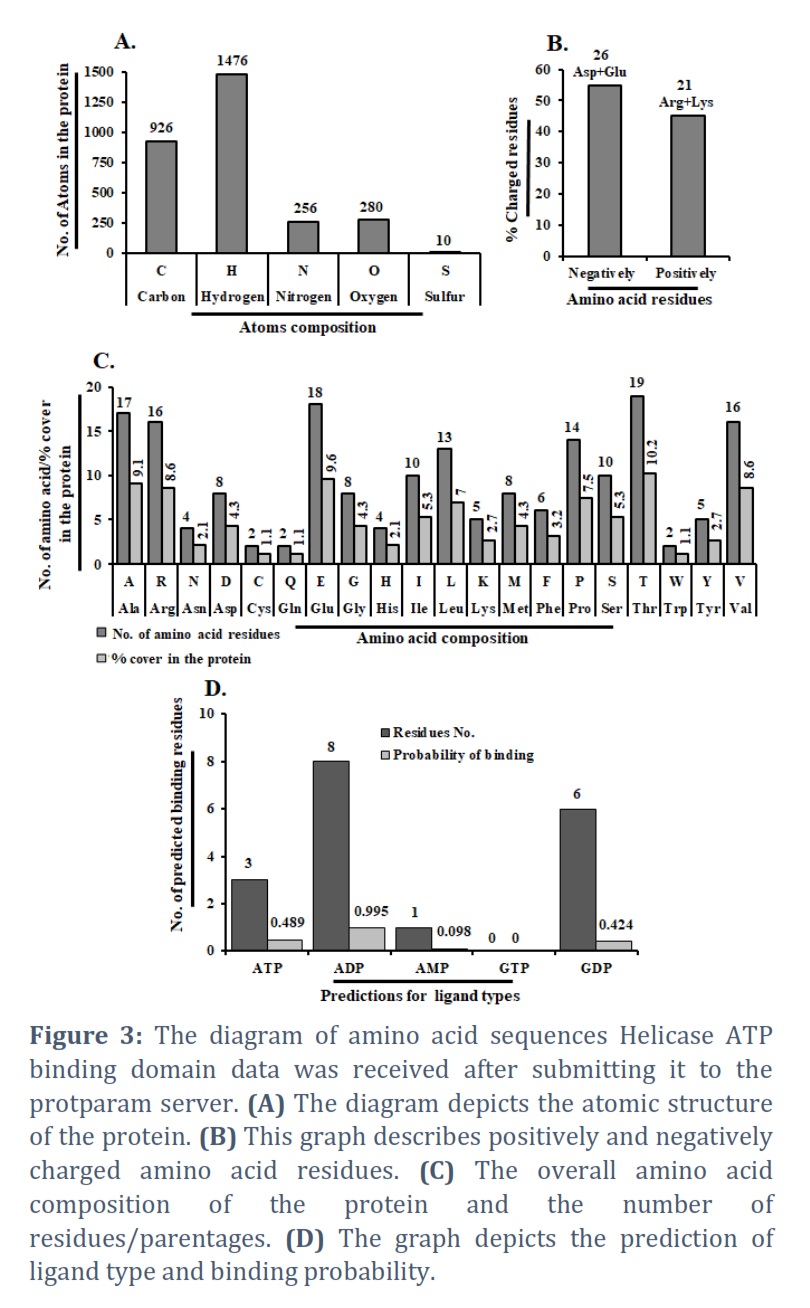
A 187-amino acid protein sequence, including the Zika virus helicase ATP binding domain. The Protparam server was used to generate the amino acid sequence. We discovered the helicase ATP binding protein’s atomic composition and several atoms. There are five atoms in total: nitrogen, sulfur, hydrogen, carbon, and oxygen. The graphical representation shows 1476 hydrogen atoms, 926 carbon atoms, 280 oxygen atoms, 256 nitrogen atoms, and ten sulfur atoms throughout the protein (Figure 3A). Finally, there are 2948 atoms in total. The hydrogen atom is essential in the composition of the helicase ATP binding domain protein, whereas sulfur plays a minor influence in its design. Similarly, we estimated the negatively and positively charged residues of this protein. We discovered that negatively charged amino acids (Asp + Glu) have a total of 26 and positively charged amino acids (Arg + Lys) have a capacity of 21 (Figure 3B). The molecular weight (20996.05) and theoretical Pi (5.46) were discovered. The entire amino acid content and several residues/parentages across the HABD protein are depicted in Figure 3C. Ala: 9.1%, Arg: 8.6%, Asn: 2.1%, Asp: 4.3%, Cys: 1.1%, Gln: 1.1%, Glu: 9.6%, Gly: 4.3%, His: 2.1%, Ile: 4.3%, Leo: 7.0%, Lys: 2.7%, Met: 4.3%, Phe: 3.2%, Pro:7.5%, Ser: 5.3%, Thr: 10.2%, Trp: 1.1%, Tyr: 2.7%, Val: 8. We also calculated the number of amino acids which are present in the HABD protein, such as A (17), R (16), N (4), D (8), C (2), Q (2), E (18), G (8), H (4), I (10), L (13), K (5), M (8), F (6), P (14), S (10), T (19), W (2), Y (5) and V(16) (Figure 3C). The graph displays the residue number and the query sequence, respectively, followed by the results of predictions for five ligand types: ATP, ADP, AMP, GDP, & GTP. Several predicted binding residues for HABD protein are ATP: 3, ADP: 8, AMP: 1, GTP: 0 & GDP: 6 (Figure 3D). Predictions for each ligand type include annotation of predicted binding residues & prediction scores, which estimate the probability of binding to a given ligand.
Identifying the phosphorylation location sites
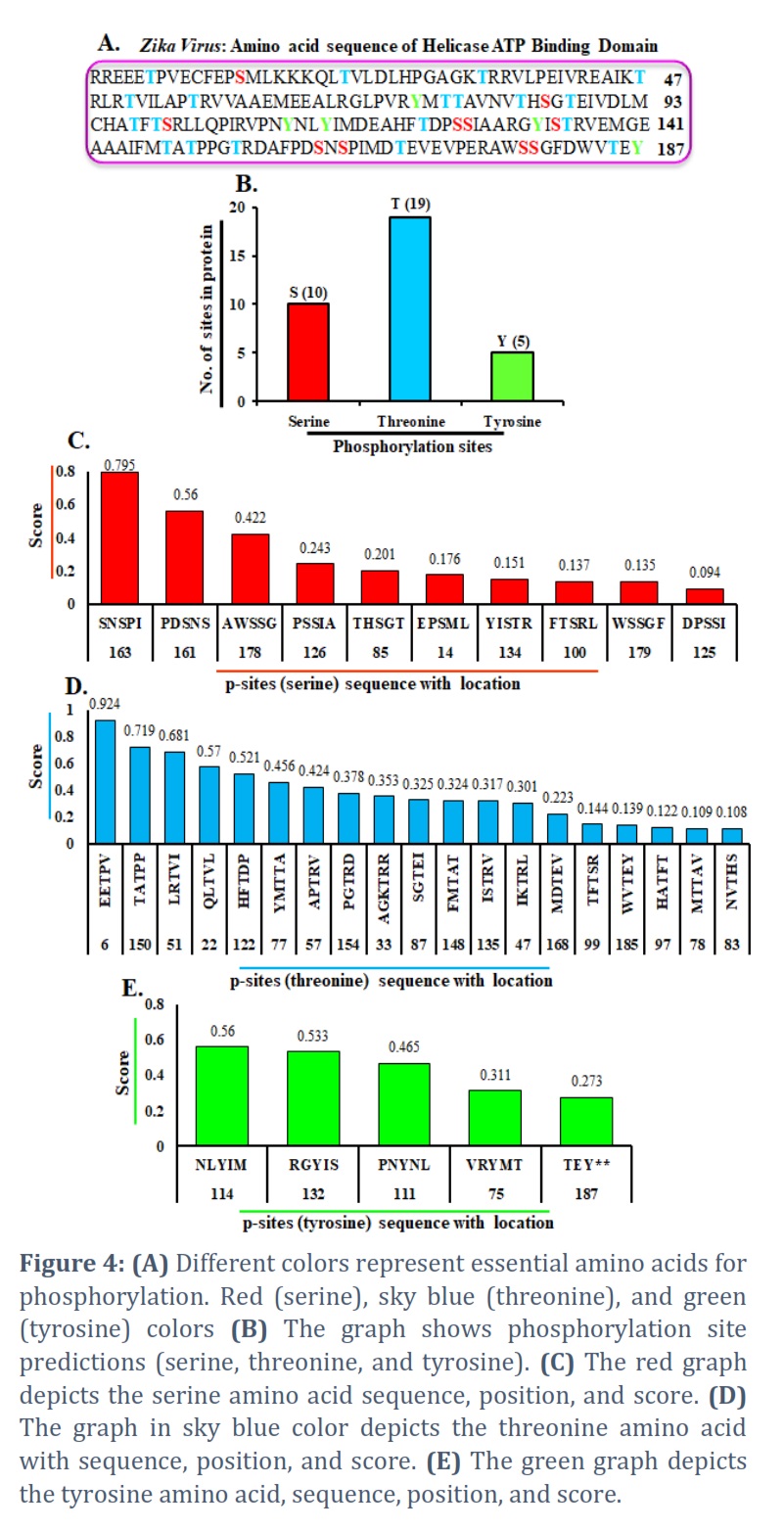
In addition, we discovered a phosphorylation site in the HABD protein sequence. A protein sequence & kinase type are required as input to kinase prediction software. Because the Helicase ATP binding domain has 187 amino acids, it generates some measure of the probability that each S/T/Y residue in the series is phosphorylated through the chosen kinase [45, 46, 47, 48]. In this sequence, we highlighted critical amino acids for phosphorylation with different colors. The red amino acid color represents the serine phosphorylation site, the threonine phosphorylation site by the sky-blue color, & the tyrosine phosphorylation site by the green amino acid color (Figure 4A). On behalf of the number of phosphorylated serine, threonine, & tyrosine amino acids in the HABD protein. Figure 4B shows a graph of the protein’s anticipated phosphorylation sites (p-sites) and several p-sites. The red color graph was prepared separately: serine, threonine, & tyrosine, with sequence, location, & scores. We discovered serine at 14, 85, 100, 125, 126, 134, 161, 163, 178, & 179 sites with scores of 0.176, 0.201, 0.137, 0.094, 0.243, 0.151, 0.56, 0.795, 0.422, & 0.135 (Figure 4C).
Similarly, with threonine, we created a sky-blue color graph with sequence, position, & scores. We found that threonine was present at 6, 22, 33, 47, 51, 57, 77, 78, 83, 87, 97, 99, 122, 135, 148, 150, 154, 168, 185, & locations with scores 0.924, 0.57, 0.353, 0.301, 0.681, 0.424, 0.456, 0.109, 0.108, 0.325, 0.122, 0.144, 0.521, 0.317, 0.324, 0.719, 0.378, 0.223, & 0.139 respectively (Figure 4D). We continuously show a green color plotted graph for tyrosine sequence, location, & scores. The results show that tyrosine is present at 75, 111, 114, 132, & 187, corresponding scores of 0.311, 0.465, 0.56, 0.533, and 0.273 (Figure 4E). Finally, we presented serine, threonine, and tyrosine p-sites and their sequence, position, & score.
GPS 6.0 is a group-based prediction system for envisaging PK-specific phosphorylation sites (p-sites)
Protein phosphorylation is one of the most researched posttranslational modifications (PTMs) that affect biological processes [49]. We used ten-fold cross-validation to test the concert of GPS 6.0 & each feature on pS/pT & pY sites for general p-site prediction. The sequences of the input can be submitted in FASTA format.
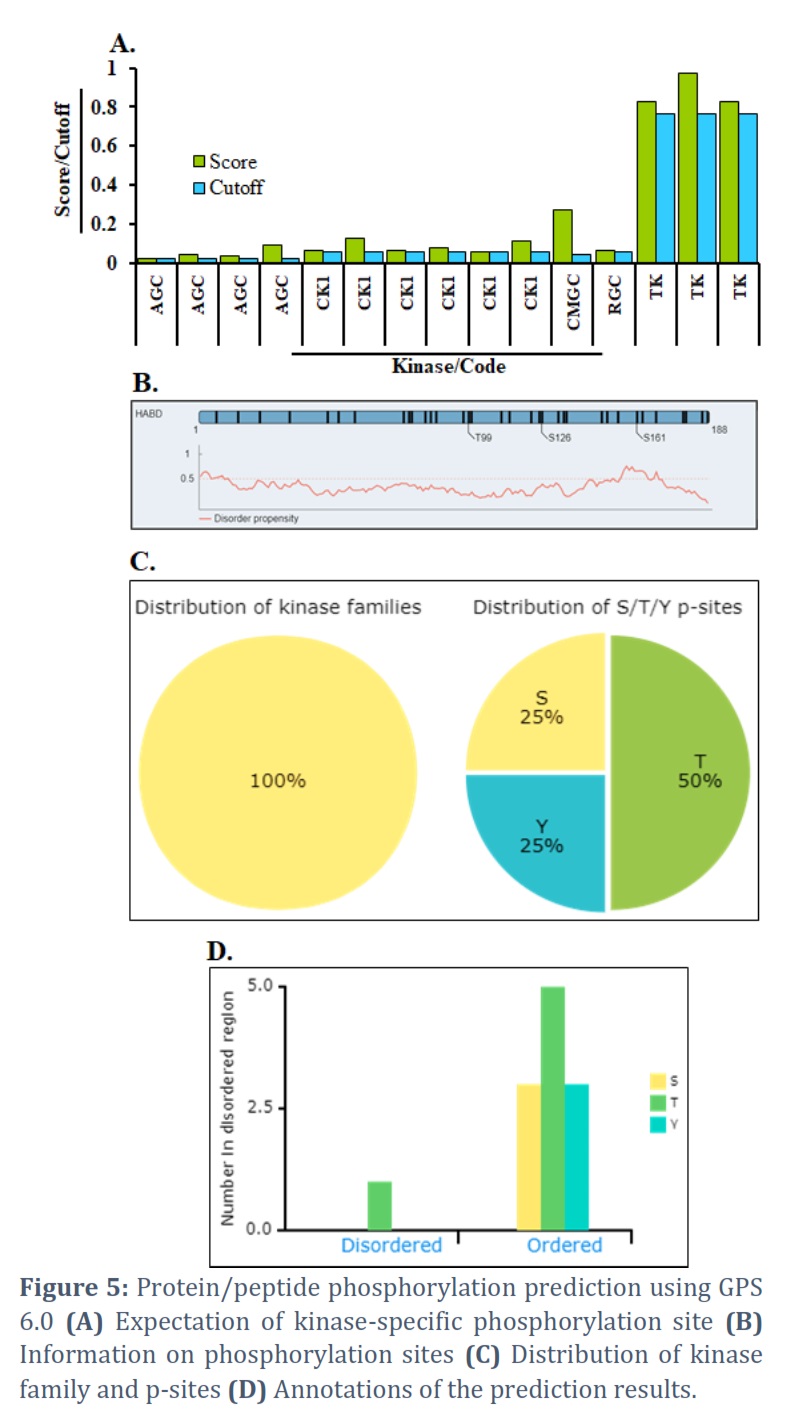
The expectation result provides possible p-sites by way of 11 different pieces of information, such as ID (the name/id of the protein sequence that you entered to forecast), Position (the location of the predicted phosphorylation site), (The residue that is expected to be phosphorylated), Kinase (the regulatory Kinase that is thought to phosphorylate the site), Score (The value calculated by the GPS
6.0 algorithm to assess phosphorylation potential. The greater the number, the more potentially phosphorylated the residue is), Cutoff (The cutoff value below the threshold. Different thresholds result in varying accuracy, sensitivity, and specificity) (Figure 5A). Surface accessibility and secondary structures, including -helix, -strand, & coil anticipated, could also be shown by NetSurfP [50] in the comprehensive mode, and they were established under the schematic figure (Figure 5B). We also ran basic statistics on the distribution of chosen kinase families, the number of disordered regions, & the secondary structures of anticipated p-sites (Figure 5C). All put in proteins’ prediction results could be taken in one of four file formats: .txt, .csv, .tsv, & .xlsx. The ‘Export’ button assists in obtaining the annotation images.png file. A help page by way of an example input & output is also given (Figure 5D).
Figures & Tables
After being isolated from Uganda for the first time in 1947, ZIKV eventually spread to other nations and regions. ZIKV has experienced several mutations and collected a variety of alterations in both structural and non-structural proteins, much like other RNA viruses [51]. Conversely, ZIKV strains that are circulating in Asian nations have the 139S residue in the preM protein, which has been linked to ZIKV-related microcephaly on occasion [52]. One element of the ZIKV replication complex is NS3. Strong contacts between the N-terminal of NS3 and the hydrophilic portion of the integral membrane protein NS2B bind it in the extracellular space. The helicase domain of the NS3 C-terminal domain is known to have NTPase and helicase activity.
The N-terminal and its cofactor NS2B combine to generate a protease that cleaves the flavivirus polyprotein. ZIKV NS3 helicase, part of the helicase superfamily 2 (SF2), is the ZIKV helicase/NTPase domain. It functions with NS5 polymerase to unwind organized template sections of RNA during the de novo viral RNA synthesis process. In addition, RNA binding stimulates the nucleoside triphosphatase activity of NS3 helicase, which supplies the chemical energy required to unwind RNA intermediates during viral RNA replication [53, 54]. They were observed when the GDP and GTP models accurately predicted the residues that bind GTP and GDP, respectively. This implies that among the predictions produced by NsitePred, structural similarity between the ligands—which influences similarity in their binding is also noted. However, the recall is significantly lower when a particular model is used to predict binding residues for less comparable nucleotides, such as when NsitePred_ATP predicts the GDP, GTP, and AMP-binding chains. This suggests that variations in nucleotide structures lead to variations in the related prediction models, which in turn drives the creation of consensus-based predictors [55]. In this study, GPS 6.0 provides PK-specific p-site predictions for 44 046 PKs across 185 species that are believable. With this assistance, more users could forecast p-sites and upstream kinases on substrates in the relevant study fields. To interpret the p-sites and substrates, we additionally integrated a lot of visible annotations. The prediction results would be given more thoroughly with the p-site and substrate information included. A user-friendly web server with highly adjustable ssKSRs prediction, selectable kinase, and various modules, GPS 6.0, was built [56]. The current study uses GPS 6.0 for amino acid composition, phosphorylation sites, genome analysis, and structure prediction.
In tropical & subtropical areas, the Aedes genus mosquito, mainly Aedes aegypti, is the principal vector for transmitting the Zika virus. The highest times for Aedes mosquito bites throughout the day are in the early morning & late afternoon, and early evening. The mosquito that spreads dengue, chikungunya, & yellow fever is the same. Using in-silico genome prediction, other domain organizations. We concentrate on the NS3 domain, the only domain with helicase unwinding and ATP binding capabilities. Additionally, we forecast helicase ATP-binding domain epitope-binding peptides. In parallel, using the Ramachandran plot, we performed structure modeling to validate. We have performed atom-composition studies of proteins, including positively and negatively charged amino acids, to gain insight into the in-depth study. We have also predicted the protein’s phosphorylation sites (p-sites) and numerous p-sites. In the future, more results-interpreting annotations will be gathered from further open sources & tools. Additionally, we’ll add more improvements to boost performance. For academic research, GPS 6.0 will be uninterruptedly sustained & amended.
Conflict of Interest
The authors declare that there is no conflict of interest.
AS and NA, prepare the first draft, wrote the MS.
![]() References
References
- Kuno G, Chang GJ, Tsuchiya KR, Karabatsos N, Cropp CB. Phylogeny of the genus Flavivirus. Journal of Virology, (1998); 72(1): 73-83.
- Centers for Disease Control and Prevention (CDC). Oseltamivir-resistant 2009 pandemic influenza A (H1N1) virus infection in two summer campers receiving prophylaxis-North Carolina, 2009. Morbidity and Mortality Weekly Report, (2009); 58 (58); 969-972.
- Zanluca C, Melo VC, Mosimann AL, Santos GI, Santos CN, Luz K. First report of autochthonous transmission of Zika virus in Brazil. Memórias do Instituto Oswaldo Cruz, (2915); 110 (4): 569-572.
- Hamelin ME, Baz M, Abed Y, Couture C, Joubert P, Beaulieu E, Bellerose N, Plante M, Mallett C, Schumer G. et al. Oseltamivir-resistant pandemic A/H1N1 virus is as virulent as its wild-type counterpart in mice and ferrets. PLOS Pathogens, (2010); 6 (7): e1001015.
- Baz M, Boivin G. Antiviral Agents in Development for Zika Virus Infections. Pharmaceuticals (Basel), (2019); 12(3):101.
- Luo D, Xu T, Hunke C, Gruber G, Vasudevan SG, Lescar J. Crystal structure of the NS3 protease-helicase from dengue virus. Journal of Virology, (2008); 82(1):173-183.
- Van Regenmortel MH. The concept and operational defnition of protein epitopes. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, (1989); 323(1217):451-466.
- Walker JM. The Proteomics Protocols Handbook. Humana press, New Jersey.(2005).
- Van Regenmortel MH. Pitfalls of reductionism in the design of peptide based vaccines. Vaccine, (2004); 19 (17-19):2369-2374.
- Serman TM, and Gack MU. Evasion of Innate and Intrinsic Antiviral Pathways by the Zika Virus. Viruses, (2019); 11(10): 970.
- Mann M, Ong SE, Gronborg M, Steen H, Jensen ON, and Pandey A. Analysis of protein phosphorylation using mass spectrometry: deciphering the phosphoproteome. Trends in Biotechnology, (2002); 20 (6): 261-268.
- Bilbrough T, Piemontese E, Seitz O. Dissecting the role of protein phosphorylation: a chemical biology toolbox. Chemical Society Review, (2022); 51(13): 5691-5730.
- Johnson SA, Hunter T. Kinomics: methods for deciphering the kinome. Nature Methods, (2005); 2(1): 17-25.
- Sharma K, Souza RC, Tyanova S, Schaab C, Wisniewski JR, Cox J, Mann M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Reports, (2014);
- 8 (5): 1583-1594.
- Zhou FF, Xue Y, Chen GL Yao X. GPS: a novel group-based phosphorylation predicting and scoring method. Biochem. Biochemical and Biophysical Research Communications, (2004); 325 (4): 1443-1448.
- Xue Y, Zhou F, Zhu M, Ahmed K, Chen G, Yao X. GPS: a comprehensive www server for phosphorylation sites prediction. Nucleic Acids Research, (2005); 33, W184-W187.
- Xue Y, Ren J, Gao X, Jin C, Wen L, Yao X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics, (2008); 7 (9): 1598-1608.
- Xue Y, Liu Z, Cao J, Ma Q, Gao X, Wang Q, Jin C, Zhou Y, Wen L, Ren J. GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Engineering, Design and Selection, (2011); 24(3): 255-260.
- Liu Z, Yuan F, Ren J, Cao J, Zhou Y, Yang Q, Xue Y. GPS-ARM: computational analysis of the APC/C recognition motif by predicting D-boxes and KEN-boxes. PLoS One,(2012); 7 (3): e34370.
- Tarique M, Ahmad M, Chauhan M, Tuteja R. Genome wide in silico analysis of the mismatch repair components of Plasmodium falciparum and their comparison with human host. Frontiers in microbiology, (2017); 8:130.
- Afaq S, Atiya A, Malik A, Alwabli AS, Alzahrani DA, Al-Solami HM, Alzahrani O, Alam Q, Kamal MA, Abulfaraj AA et al. Analysis of methyltransferase (MTase) domain from Zika virus (ZIKV). Bioinformation, (2002); 16(3):229-235.
- Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL. The Pfam protein families database. Nucleic acids research,(2002); 30(1):276-280.
- Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Research, (2006); 2(2):2.
- Wu S, Skolnick J, Zhang Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC biology, (2007); 5: 17.
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols, (2010); 5 (4): 725-738.
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. Journal of molecular biology, (1999); 292 (2): 195-202.
- Wu S, Zhang Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic acids research, (2007); 35 (10): 3375-3382.
- Zhang Y, Kihara D, Skolnick J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins: Structure, Function, and Bioinformatics, (2002); 48 (2): 192-201.
- Wu S, Zhang Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics, (2008); 24 (7): 924-931.
- Zhang Y, Skolnick J. SPICKER: A clustering approach to identify near‐native protein folds. Journal of computational chemistry, (2004); 25 (6): 865-871.
- Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic acids research, (2005); 33(7): 2302-2309.
- Li Y, Zhang Y. REMO: A new protocol to refine full atomic protein models from C‐alpha traces by optimizing hydrogen‐bonding networks. Proteins: Structure, Function, and Bioinformatics, (2009); 76 (3): 665-676.
- Lovell SC, Davis IW, Arendall WB, de Bakker PI, Word JM, et al. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins: Structure, Function, and Bioinformatics, (2003); 50 (3): 437-450.
- Lawrence A Kelley, Stefans Mezulis, Christopher M Yates, Mark N Wass & Michael J E Sternberg. The Phyre2 web portal for protein modeling, prediction and analysis. Nature Protocols, (2015); 10 (6):845-858.
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein. Identification and Analysis Tools on the ExPASy Server; (In) John M. Walker (ed): The Proteomics Protocols Handbook, Humana Press, (2005); 571-607.
- Boeckmann B, Bairoch A, Apweiler R, et al. The Swiss-Prot protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Research, (2003); 31 (1): 354-370.
- Chen K, Mizianty MJ, Kurgan L. Prediction and analysis of nucleotide-binding residues using sequence and sequence-derived structural descriptors. Bioinformatics, (2012); 28 (3): 31-34.
- Dou Y, Yao B, Zhang C. PhosphoSVM: Prediction of phosphorylation sites by integrating various protein sequence attributes with a support vector machine. Amino Acids, (2014); 46(6):1459-69.
- Ahmad S, Gromiha MM, Sarai A. RVP-net: online prediction of real valued accessible surface area of proteins from single sequences. Bioinformatics, (2003); 19(14):1849-1851.
- Basu S, Plewczynski D. AMS 3.0: prediction of post-translational modifications. BMC Bioinforma, (2010); 11:210.
- Chen M, Zhang W, Gou Y, Xu D, Wei Y, Liu D, Han C, Huang C, Li C, Ning W, Peng D, Xue Y. GPS 6.0: an updated server for prediction of kinase-specific phosphorylation sites in proteins, Nucleic Acids Research, (2023); 51(W1): W243-W250.
- Wang C, Xu H, Lin S, Deng W, Zhou J, Zhang Y, Shi Y, Peng D, Xue Y. GPS 5.0: An update on the prediction of kinase-specific phosphorylation sites in proteins. Genomics, Proteomics & Bioinformatics, (2020); 18(1):72-80.
- Xue Y, Liu Z, Cao J, Ma Q, Gao X, Wang Q, Jin C, Zhou Y, Wen L. Jian Ren. GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Engineering, Design and Selection, (2011); 24 (3): 255-260.
- Baz M, and Boivin G. Antiviral Agents in Development for Zika Virus Infections. Pharmaceutics, (2019); 29 12(3):101.
- Si-Qiao TAN, Qian LI, Yuan C, Jian P. Phosphorylation Site Prediction Integrating The Position Feature With Sequence Evolution Information. Progress in Biochemistry and Biophysics, (2017); 44(12): 1118-1124.
- Pinna LA, Ruzzene M. How Do Protein Kinases Recognize Their Substrates? Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, (1996); 1314 (3): 191-225.
- Newman RH, Zhang J, Zhu H. Toward a Systems-Level View of Dynamic Phosphorylation Networks. Frontier in Genetics, (2014); 15:5:263.
- Trost B and Kusalik A. Computational Prediction of Eukaryotic Phosphorylation Sites. Bioinformatics, (2011); 27(21):2927-35.
- Mann M, Ong SE, Gronborg M, Steen H, Jensen on and Pandey A. Analysis of protein phosphorylation using mass spectrometry. Trends in Biotechnology, (2002); 20(6):261-8.
- Petersen B, Petersen TN, Andersen P, Nielsen M and Lundegaard C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Structural Biology, (2009); 9: 51.
- Aziz A, Suleman M, Shah A, Ullah A, Rashid F, et al. Comparative mutational analysis of the Zika virus genome from different geographical locations and its affect on the efficacy of Zika virus-specific neutralizing antibodies. Frontiers in Microbiology, (2023); 22:14:1098323.
- Wongsurawat T, Athipanyasilp N, Jenjaroenpun P, Jun SR, Kaewnapan B, Wassenaar TM. et al. Case of microcephaly after congenital infection with Asian lineage Zika virus, Thailand. Emerging Infectious Diseases, (2018); 24:1758.
- Luo D, Vasudevan SG, Lescar J. The flavivirus NS2B-NS3 protease-helicase as a target for antiviral drug development. Antiviral Research, (2015); 118:148-158.
- Tay MYF, Saw WG, Zhao Y, Chan KWK, Singh D, Chong Y, et al. The C-terminal 50 amino acid residues of dengue NS3 protein are important for NS3-NS5 interaction and viral replication. Journal of Biological Chemistry, (2015); 290(4):2379-2394.
- Ke Chen, Marcin J, Mizianty and Kurgan L Prediction and analysis of nucleotide-binding residues using sequence and sequence-derived structural descriptors. Structural bioinformatics, (2012); 28(3): 331-341.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0