

Full Length Research Article
Target-based virtual screening and molecular dynamics approach to identify potential antileishmanial agents through targeting UvrD-like helicase ATP-binding domain
Misbahuddin M Rafeeq1, Nawal Helmi2, Ziaullah M Sain3, Johar Iqbal4, Abdulrahman Alzahrani5, Mohammad Othman Alkurbi6, Abdullah F. Shater7, Bassam M. Al-ahmadi8, Mohammad Zubair Alam9,10, Qamre Alam11*
Adv. life sci., vol. 11, no. 1, pp. 237-245, February 2024
*– Corresponding Author: Qamre Alam (alamqa2022@gmail.com)
Authors' Affiliations
2. Department of Biochemistry, College of Science, University of Jeddah, Jeddah – Saudi Arabia
3. Department of Microbiology, Faculty of Medicine, Rabigh. King Abdulaziz University, Jeddah- Saudi Arabia
4. Department of Biochemistry, Faculty of Medicine, Jazan University – Saudi Arabia
5. Department of Applied Medical Sciences, Applied College, Al-Baha University, Al-Baha City – Saudi Arabia
6. Department of Clinical Laboratory Sciences, Faculty of Applied Medical Sciences, Umm Al-Qura University, Makkah – Saudi Arabia
7. Department of Medical Laboratory Technology, Faculty of Applied Medical Sciences, University of Tabuk, Tabuk – Saudi Arabia
8. Department of Biology, Faculty of Science, Taibah University – Saudi Arabia
9. Clinical Research Unit, King Fahd Medical Research Center, King Abdulaziz University, Jeddah – Saudi Arabia
10. Department of Medical Laboratory Technology, Faculty of Applied Medical Sciences, King Abdulaziz University, Jeddah – Saudi Arabia
11. Molecular Genomics and Precision Medicine Department, ExpressMed Diagnostics and Research, Block – Kingdom of Bahrain
[Date Received: 29/07/2023; Date Revised: 29/08/2023; Date Published: 25/02/2024]
Abstract![]()
Introduction
Methods
Results
Discussion
References
Abstract
Background: About 0.7-1.0 million people worldwide have been suffering from Leishmaniasis. It falls under a neglected tropical disease (NTD) and is transmitted by biting infected female phlebotomine sandflies. The implication of “the NTD road map: together towards 2030” in the infection-prone regions worldwide has curtailed morbidity to a greater extent. However, limited options in antileishmanial oral and topical drugs must decipher more therapeutically efficacious agents to cure and eradicate the disease.
Methods: Virtual screening based on structure, docking, & molecular dynamics approaches were adopted to identify potential lead molecules against UvrD-like helicase of Leishmania donovani from the MCULE database. Lipinski rule of five, N/O atoms (1-15), number of rings (1-2), HBDs (4-5), and HBAs (5-10) were applied as initial filters of SBVS. AutoDock Vina and GROMACS packages were used for docking and MD simulations, respectively.
Results: Initial filters of SBVS workflow yielded 93885 ligand hits out of 100 plus million investigational ligands. Following the toxicology test, 28 ligands were gotten that were additional reduced to molecules (17) when accepted done the BOILED Egg model of the ADME. Six molecules were shortlisted with zero violation compliance of drug-likeness further than Lipinski RO5 viz., Egan, Veber, Muegge, Ghose, & bioavailability score having ΔG (-6.7 to -7.4 kcalmol-1) lesser than reference inhibitor miltefosine (-4.9 kcalmol-1). The stability of MCULE-5754880195-0-2 was found to be greater than the known inhibitor and ligand molecules mentioned above.
Conclusion: MCULE-5754880195-0-2 has all therapeutic features by way of an admirable oral drug molecule & could be encouraging in Leishmaniasis prevention & treatment.
Keywords: UvrD-like helicase; ADME; Leishmaniasis; MCULE database; SBVS; Docking; BOILED Egg; MD simulation; ATP-binding domain
Introduction![]()
Leishmaniasis, which is regarded as one of the most neglected tropical diseases, is thought to be caused by more than twenty different Leishmania species. Over ninety sandfly species are known to spread Leishmania parasites. About 0.7-1.0 million new cases occur annually worldwide, especially in socially and financially deprived countries, although the progression of infection towards developing leishmaniasis is slow [1]. Three types of leishmaniasis exist. viz., visceral, mucocutaneous & cutaneous. The first one, visceral leishmaniasis (VL), is the fatal form of the disease, categorized via asymmetrical attacks of expansion of the spleen & liver, weight loss, fever, & anemia. The World Health Organization (WHO) claims, 2020 statistics >90% of VL incidence were reported from Brazil, Somalia, Yemen, Sudan, India, China, Ethiopia, Eritrea, Kenya, & South Sudan [2]. The second one is CL (cutaneous leishmaniasis) the utmost public form of the disease that causes severe skin wounds and scars mostly on the uncovered parts of the body. America, Mediterranean, Middle East, & Central Asia contribute over 90% of CL incidences compared to other countries [3]. The third one, mucocutaneous leishmaniasis (ML), is a comparatively mild form of the disease that destroys the membranes (mucous) of the nose, mouth, & throat. Maximum cases of ML arise in Brazil, Bolivia, Peru, & Ethiopia [4]. Leishmania parasites are transmitted when female phlebotomine sandflies infected suck the blood of the reservoir host to lay eggs. Several characteristics, viz., ecology of transmission sites, vector species, and present & past contact of the human populace to the parasite, influence the epidemiology of leishmaniasis. Besides humans, > 65 species (animal) have been reported as normal reservoir hosts of the leishmaniasis-causing organism [5]. People suffering from both leishmaniasis, and HIV are more prone to full blown medical disease & extraordinary revert & mortality rates, often seen in Ethiopia, Brazil, & India (Bihar) [6]. Moreover, socioeconomic conditions, environmental changes, population mobility, malnutrition, & climate change are the key risk factors associated with leishmaniasis. Prevention and control of leishmaniasis require holistic strategies because spreading the disease occurs through the complex biological system of the host revisor human, causing agents and vectors. Albeit persisting complexities in treatment options, primary diagnosis & effective quick treatment, effective disease observation, regulator of animal reservoir hosts, vector control, community mobilization, & consolidation partnerships are a few critical strategies for prevention and control of leishmaniasis. Moreover, the WHO’s subsidized medicine price and donation program have accelerated disease prevalence reduction, curtailing the associated disability and mortality to a certain extent [7].
According to classical concepts, the pathogenesis of leishmaniasis arises due to the inequity of T helper type 1 & T helper type 2 (TH1 & 2) cells [8]. Many complexities persist with the diagnosis of Leishmaniasis. Two approaches, direct (parasitic) and indirect (immunological), are standard for diagnosis. However, confirmatory inferences are not so easy in impoverished populations. Despite various problems, clinicians diagnose in the best way based on the history, epidemiology, clinical symptoms, and physical appearance of scars and lesions. There is limited oral therapeutics for the treatment of Leishmaniasis. Miltefosine and a few azoles viz., Fluconazole and Ketoconazole are used in used in systemic therapy. Miltefosine (DB09031) is the sole recognized oral drug to treat VL, CL, and ML. However, it was developed in 1980 as an anticancer agent, but now it has been repurposed as a broad-spectrum antileishmanial, antimicrobial, & phospholipid drug. Patients above the age of 12 are given it orally or topically. CDC (Centres for Disease Control and Prevention) has also suggested this one as a first line treatment for
Methods![]()
Structure (3D) prediction & optimization of target protein
Swiss-Model web tool was used to 3D structure Leishmania donovani (strain BPK282A1). The model was built based on the template DNA helicase (PDB ID: 1PJR) of Geobacillus stearothermophilus (UniProt ID: P56255) [16]. Qualitative Model Energy Analysis (QMEAN) tool using consensus-based distance constraint, a.k.a. QMEANDisCo, and Ramachandran plot was used to evaluate the qualitative and stereochemical quality for the predicted model [17]. The target protein was modified by applying the CHARMm force field [18,19].
SBVS (Structure Based Virtual Screening)
MCULE, an online drug discovery platform, was employed for structure based virtual screening of experimental small ligands similar to miltefosine from its vast digital repository, which contains over 100 million synthetically accessible and purchasable molecules. In the structure based virtual screening (SBVS) work procedure, the fundamental search limit complying with Pfizer’s RO5, number of nitrogen and oxygen atoms, and number of aromatic rings was applied to screen chemical hits. Input values for the diversity range, sample size, & resemblance search threshold were 1000, 100, and 0.90, respectively. Small molecule fragments based robust 2D search algorithm, a.k.a. FP2 fingerprint of open babel, was assigned to execute the SBVS workflow [20,21].
Retrieval of structure (3D) &customization of reference drug
The structure (2D) of reference drug miltefosine (CID Number: 3599) in standard data format (SDF) was extracted from the database (NCBI & PubChem) [22, 23]. 2D to the 3D conversion of miltefosine was supported via the Biovia discovery studio visualizer (DSV). The ligand was actively reduced through a similar protocol by way of the target protein ULH [18, 24].
Molecular docking with AutoDock Vina
The AutoDock Vina (ADV) intrinsic toward the MCULE database (https://mcule.com/) was used for molecular docking concerning the target protein & Structure Based Virtual Screening (SBVS) – chemical hits. The PDB structure of the protein as an input file was delivered near the ADV edge of the portal (MCULE. Grid size in x (7.184 Å), y (31.4135 Å), and z (7.224 Å) directions were applied to shield the binding pockets of protein. The ADV settings (parameter) for binding mode per ligand & exhaustiveness were left at their default values. The free energy of binding (ΔG) was regarded as the most imperative factor for determining the optimal posture of ligands docked into the binding cavity of ULH [11, 25-27].
Toxicity investigation of virtually-screened hits
The existence of toxic moieties, fragments, & substructures in effectively open out ligand molecules unwelcomed in the human & eco-friendly environments was examined over and done with the Toxicity Checker tool of the MCULE database grounded on the difficult & robust SMARTS (SMILES (simplified molecular-input line-entry system) arbitrary target specification) algorithm [20].
Egan’s BOILED-Egg filtration
The BOILED (Brain or Intestinal Estimate D) Egg model of the SwissADME implement was working to predict the BBB & HIA permeation of identified hits. The BOILED Egg relies on the 2 physicochemical descriptors, viz., WLOGP (≤ 5.88 as a reference value) & TPSA (≤ 131.6 as a reference value) for lipophilicity & seeming polarity, correspondingly & distinctive illustrative explanation of exactly how faraway a molecule is from the perfect one for ideal preoccupation [28- 31].
Assessment of medicinal chemistry attributes
The medicinal chemistry properties include frequent hitters, often known as promiscuous compounds, which were anticipated using the pan assay interference structure (PAINS) observant option of SwissADME tools developed by Eli Lilly. Ruth Brenk's alert evaluated unwanted substructures, dyes, and hazardous compounds [32, 33].
Molecular dynamics (MD) simulation
GROMACS 5.1.2 was used to compute the stabilities of docked complexes of the best ligand-ULH and the reference medication miltefosine-ULH at 300K at the molecular mechanics’ level. The ligands were divided as of their corresponding docked complexes through gmx grep module. The topology and forcefield consideration (parameter) files for the selected ligand hits were predicted by the CHARMm general force field (CGenFF) programme. The topologies for ULH were created using GROMACS 5.1.2's pdb2gmx modules. The CGenFF tool was used to retrieve the structural coordinates of screened hits and the reference drug miltefosine [34, 35, 36].
By a margin of 10 Å, all ligand and reference drug guaranteed complexes were soaked in a dodecahedron box of molecules (water). The gmx editconf module was used for making boundary circumstances. Using addition chloride & sodium ions via the gmx genion module, to maintain neutrality, the charges on the bound complexes were neutralised, preserving the physiological concentration (conc.) of 0.15 M. The system was then reduced for 250000 stepladders using the sharpest pedigree algorithm. The system temperature (temp.) was elevated from 0-300 K in the course of their equilibration of 5ns interval at persistent NVT & NPT. Subsequently the equilibration phase, the particle mesh was allocated through Ewald method [37, 38]. Different modules of the GROMACS package viz., gmx rmsf, gmx rms, gmx sasa & gmx ΔGsolv, & gmx Rg, were used to depict the stability of lead molecules in terms of RMSD (root square deviation), RMSF (root mean-square fluctuation), SASA (solvent accessible surface area) the free energy of solvation (ΔGsolv), & radius of gyration (Rg) plots [34, 39].
Results![]()
Structure (3D) prediction of UvrD-like helicase, virtual screening and toxicity check
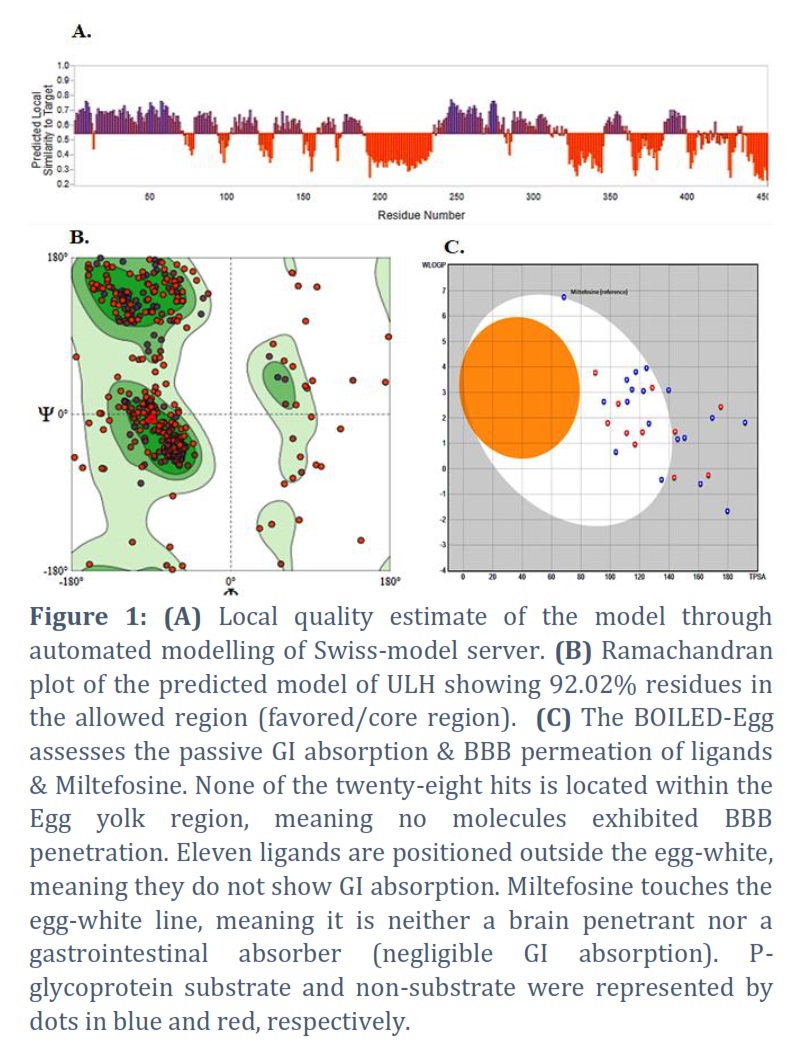
Swiss-Model server built the model of UvrD-like helicase (E9B9V1) using template DNA helicase (PDB ID: 1PJR) of Geobacillus stearothermophilus (UniProt ID: P56255) (16), which target-template sequence similarity was found to be 27.72%. Average Model Confidence in terms of QMEANDisCo global score was found to be 0.54 ± 0.05. Ramachandran plot exhibited that 92.02 % of residues of the model were located in its core region. Predicted models with >90% residues in the core region are considered promising [40]. The local confidence of the predicted model and Ramachandran plot is illustrated in Figures 1A and 1B, respectively.
Method (SBVS) was employed to identify small molecules from the MCULE’s digital source of new ligands (>100 million). Basic filters of RO5 (zero violation), number of N/O atoms (range: 1-15), and aromatic rings (limit: 1-2) yielded 93885 ligand hits, followed by toxicity assessment through the SMARTS algorithm of SwissADME. Twenty-eight ligands turned out as non-toxic drug nominees, & the remaining 93,857 ligands were disallowed during rigorous toxicity checking.
The BOILED-EGG filtering technique
For instance, the name implies, the Egan model consists of two regions, i.e., yellow & white, depicting the physicochemical spaces for considerable BBB (blood-brain barrier) permeation & gastrointestinal (GI) absorption, a.k.a. human intestinal absorption (HIA). Seventeen molecules (out of 28) showed significant BBB penetration and HIA permeation. The BOILED-Egg extrapolation of 28 ligands & reference molecules, i.e., miltefosine, is revealed in Figure 1C. The blue color & red color dots denote P-gp (+ve & P-gp –ve molecules, which means some ligands that are a substrate of P-glycoprotein have effluxed out in the course of BBB dispersion, & non-substrate ligands can penetrate the brain membrane [28, 42].
Drug-likeness other than Lipinski RO5
Drug-likeness models viz., Muegge (Bayer), Veber (GSK), Ghose (Amgen & Egan (Pharmacia) Abbott bioavailability score (BS) were used to identify pro-drug molecules [30, 31, 42, 43, 44, 45]. These models qualitatively estimate the drug-likeness for a molecule (small) to develop future potential oral prime molecules. Out of seventeen, only six ligands, MCULE-5855858205-0-1, MCULE-8153392673-0-37, MCULE-5754880195-0-2, MCULE-4495139953-0-1, MCULE-7191245305-0-1, and MCULE-4952526932-0-1 obeyed the rules mentioned above with zero violation, and eleven ligands breached the law. Miltefosine exhibited 2, 1, 1, & 2 violations of Ghose, Veber, Egan, & Muegge models, correspondingly. Moreover, all six ligands and miltefosine exhibited a similar Abbott BS of 0.55.
Docking simulation
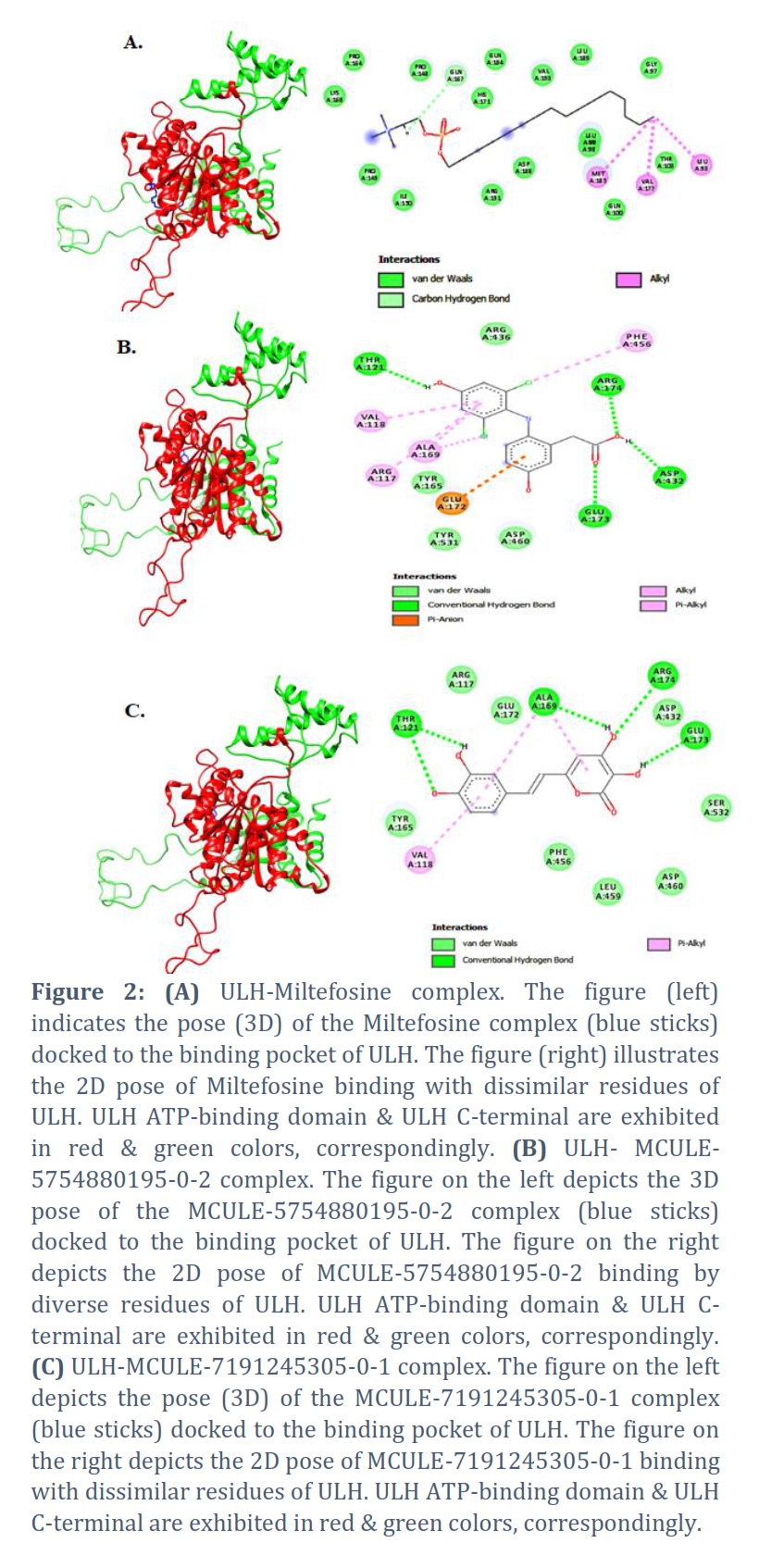
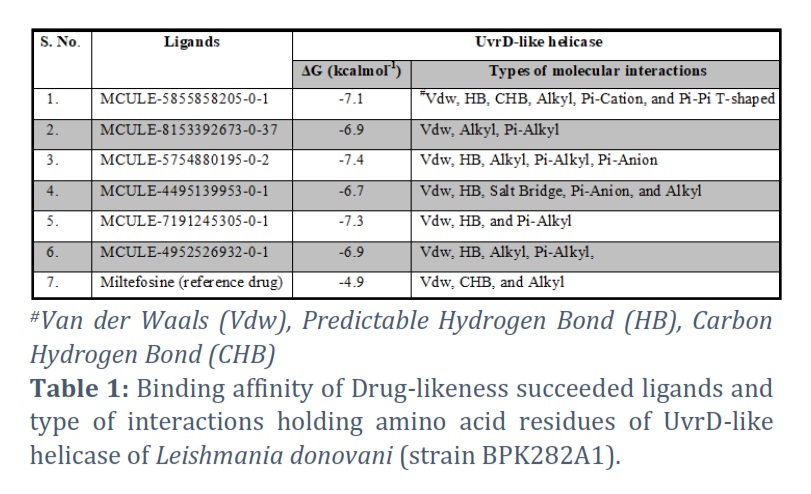
Drug-likeness Using MCULE's ADV tool, successful ligands and Miltefosine were docked into the binding cleft of protein (target) ULH to analyze their binding affinities in terms of binding free energy (ΔG) ranging from -7.4 to -4.9 kcalmol-1. Docked complexes of anticipated ligands & ULH were paralleled with reference molecule Miltefosine in terms of ΔG & type of connections. Miltefosine interacted into the binding pocket of ULH with a ΔG value of -4.9 kcalmol-1 unveiling interactions with 20 residues via three different binding interactions viz., van der Waals (Vdw), carbon-hydrogen bond & Alkyl bonds (Figure 2A). Based on the ΔG criterion MCULE-5754880195-0-2 and MCULE-7191245305-0-1 were found as the top two ligands illustrating interactions with 13 residues through five (Vdw, HB, Alkyl, Pi-Alkyl, and Pi-Anion) and three (Vdw, HB, and Pi-Alkyl) binding interactions respectively (Figure 2B-C). Binding affinity and types of interaction involved in all six ligands passing through drug-likeness filtration are shown in Table 1.
Hydrogen bond formation and Stability investigation over MD simulations
The mention (reference) drug Miltefosine revealed nil conservative hydrogen bonds through binding interaction with the aim protein ULH, however MCULE-5754880195-0-2 and MCULE-7191245305-0-1 showed 4 and 5 hydrogen bonds respectively during interaction with ULH. These two ligands displayed a strong binding affinity by more negative ΔG values with target protein residues as compared to other selected ligands and inhibitor. As a result, only compounds with the greatest number of hydrogen bonds and the least amount of ΔG were chosen for MD investigations. [46, 47]. The stability of the top ligand hit docked complexes viz., MCULE-5754880195-0-2 & reference drug Miltefosine with ULH was investigated through MD simulations of 100 ns duration at Dell Workstation Precision 3440 using GROMACS (Groningen MAchine for Chemical Simulations) package. MD graphs for SASA, RMSF, RMSD, ΔGsolv, Rg, & HBs were plotted to assess the molecular docking stability of ligands & protein docked complexes [34].
RMSD (Root mean square deviation), RMSF (Root mean square fluctuation), SASA (Solvent accessible surface area) and Free solvation energy
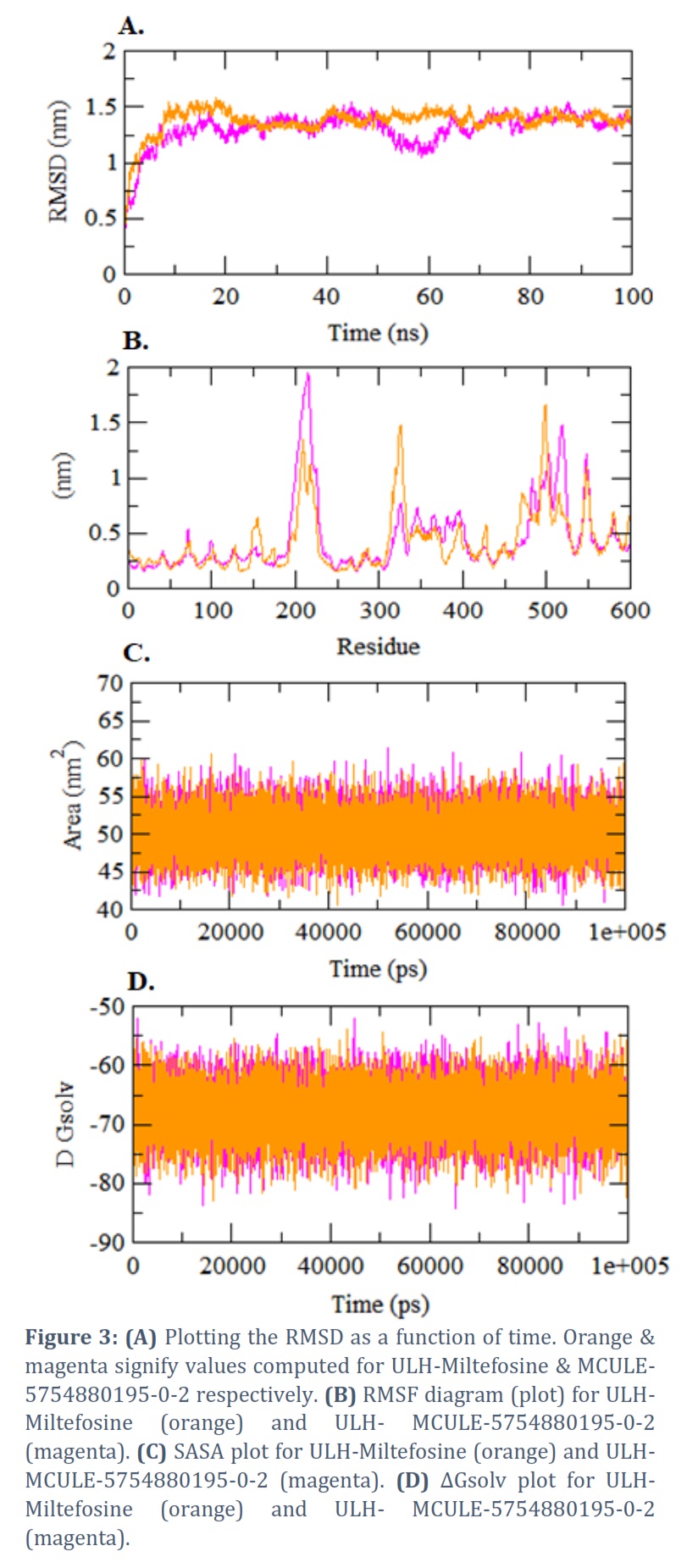
The RMSD informs us about the stability of docked complexes. The mean RMSD for mention (reference) inhibitor Miltefosine (orange) & anticipated ligand hits MCULE-5754880195-0-2 (magenta) complexed with ULH was found 1.364 nm and 1.358 nm, correspondingly. The RMSD plot conceals that the stability of docked complex of ULH & MCULE-5754880195-0-2 is comparatively than inhibitor (Figure 3A).
The residues variations at numerous areas of the RMSF diagram (plot) are due to the binding interactions of Miltefosine & MCULE-5754880195-0-2 with ULH. Average residues fluctuation upon binding through Miltefosine (orange) and MCULE-5754880195-0-2 (magenta), was establish as 1.318 nm and 0.707 nm correspondingly (Figure 3B).
The SASA plot represents the surface area of the protein that is nearby to the solvent molecule. The usual (average) SASA values upon binding with Miltefosine (orange) & MCULE-5754880195-0-2 (magenta) was found as 94.452 nm2 and 104.705 nm2 respectively (Figure 3C).
The average ΔGsolv of ULH upon binding through Miltefosine (orange) & MCULE-5754880195-0-2 (magenta) was depicted as -126.761 kJ/mol/nm2 & -144.476 kJ/mol/nm2 correspondingly (Figure 3D).
Gyration radius and HBs formation & deformation
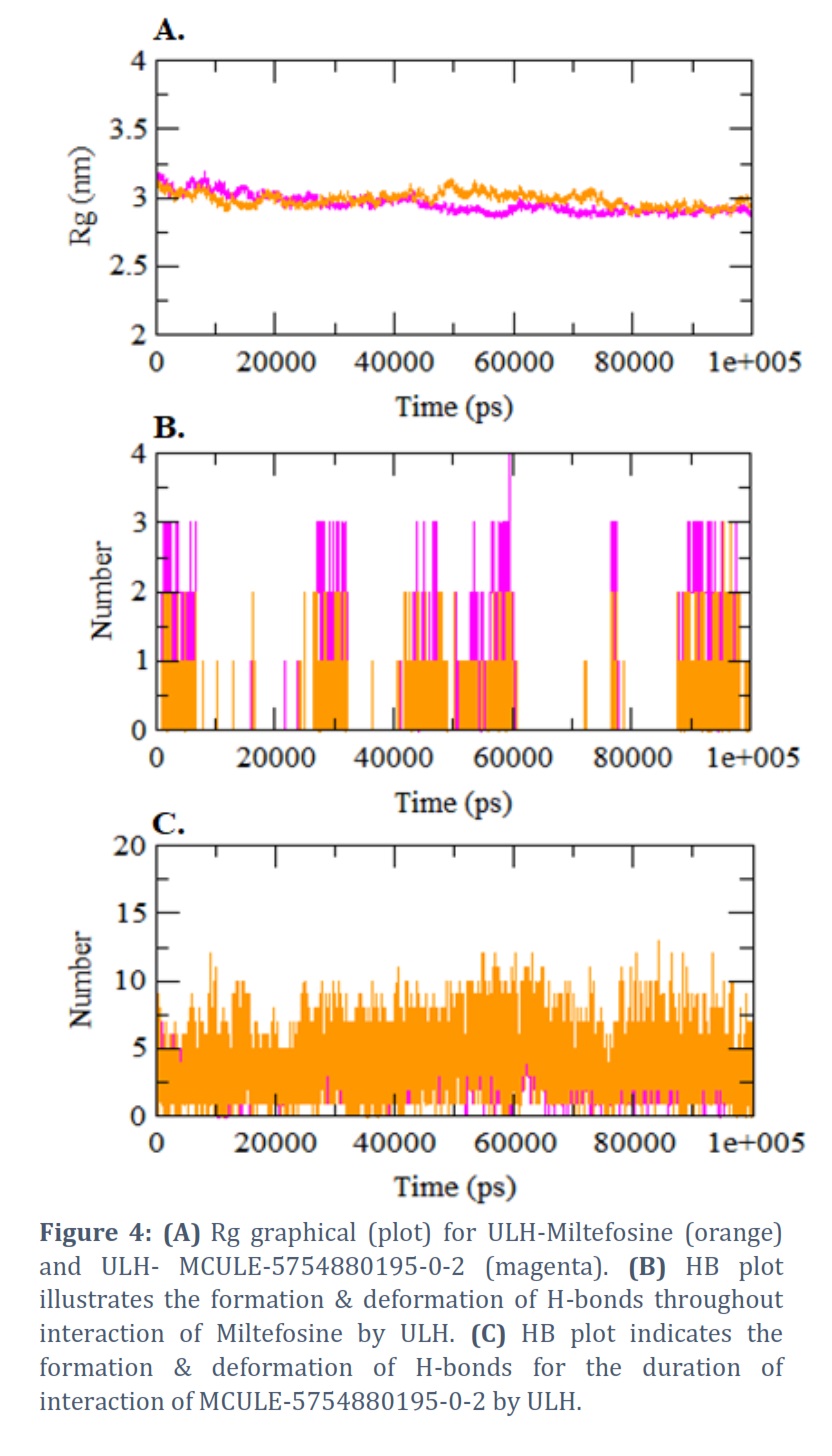
The Rg masks the density of docked complexes and is inversely related to compactness. The ordinary (average) Rg values of docked complexes of Miltefosine (orange) & MCULE-5754880195-0-2 (magenta) were set up as 6.016 nm and 5.971 nm correspondingly (Figure 4A).
The HB plot illustrates the number of hydrogen bond formations, deformation & stability during the all-inclusive process of simulations (MD).
Figure 4B indicates the HB plot for the docked complex Miltefosine-ULH and Figure 4C represents the nature of HB formation & deformation of ULH- MCULE-5754880195-0-2 during the all-inclusive course of molecular dynamics simulation of 100 ns period. Single HB is formed in ULH-Miltefosine, but they did not attain stability till the end of 50 ns MD simulation, though in the case of MCULE-5754880195-0-2, 5 HBs are being formed, that attains stability till the entire duration.
Figures & Tables
The main goal of finding strong and targeted helicase inhibitors is to regulate how an organism can access its genetic material. Helicase inhibitors could theoretically be used to regulate any aspect of gene expression or replication. Nevertheless, the majority of current research focuses on developing helicase inhibitors that stop cancer cells or infectious infections from proliferating. Strong and targeted inhibitors of bacterial helicases, like the DnaB protein that acts at bacterial replication forks, or proteins involved in recombination, like RecBCD, may be used to create antibiotics. Cellular helicase inhibitors have the potential to function as antivirals, regulate cancer cells, or increase their susceptibility to chemotherapy [48,49,50]. The creation of medications to treat leishmaniasis is moving very slowly. Leishmaniasis can be treated with a limited number of medications, but managing the disease is made more difficult by toxicity, side effects, high costs, and the emergence of drug resistance. In order to prevent the spread of resistant Leishmania parasites and to help control leishmaniasis, it is necessary to develop new hits and leads [51]. The drug design and development field has seen a revolution in recent years thanks to nanotechnology. Compared to conventional chemotherapeutic options, it has demonstrated promise as a tool in parasitic diseases where the parasite relapses. Two ways nanotechnology has facilitated drug development are the creation of drug delivery systems and the nano formulation of medications that make them easy targets for macrophage phagocytosis, resulting in targeted drug delivery [52]. Leishmania adapts in advance to the environmental conditions of the host macrophage. Autophagy, which uses cathepsins and cysteine peptidases to regulate protein turnover, is an integral part of the life cycle of parasites and an essential step in their development and differentiation. This made these molecules viable therapeutic targets for future drug development [53].
ULH of Leishmania donovani is a putative therapeutic target against which drug design could be a lucrative strategy to prevent and treat Leishmaniasis. The predicted model of ULH depicts low sequence similarity with the selected template, but its more than ninety percent (90%) residues located in the most favored area (region) of the Ramachandran plot favor the useability in computer-aided drug design. High-throughput SBVS, profiling, physicochemical, toxicity properties, solubility, lipophilicity, drug-likeness, pharmacokinetics, medicinal chemistry attributes, molecular docking, SASA, RMSF, RMSD, ΔGsolv, Rg & HBs evaluates determine that MCULE-5754880195-0-2 covers all therapeutic features that are superior to Miltefosine. In the purview of the above findings, MCULE-5754880195-0-2 may have emerged as an excellent oral drug candidate against Leishmaniasis. Experiments adopted are based on bioinformatics and computational tools having several possible research limits. Therefore, comprehensive wet-lab investigations are required to authenticate the in-silico findings of the research.
Acknowledgement
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number KEP-1-141-42 and King Abdulaziz University, DSR, Jeddah, Saudi Arabia.
Author Contributions
MMR, MZA and QA: designed and first drafted the manuscript. ZMS, JI and AA: data analysis, and revised the manuscript. MO and AFS: data collection and editing of the manuscript. BMA and NH: Final revision and editing of the manuscript. Authors read and approved the final version.
The authors declare that there is no conflict of interest.
![]() References
References
- Mishra R, Rohatgi I. (2021). Leishmaniasis. Indian Journal of Critical Care Medicine, (2021); 25(S2):S166-70.
- Van Griensven J, Diro E. Visceral Leishmaniasis. Infectious Disease Clinics of North America, (2019); 33(1):79-99.
- Abadías-Granado I, Diago A, Cerro PA, Palma-Ruiz AM, Gilaberte Y. Leishmaniasis cutánea y mucocutánea. Actas Dermosifiliog, (2021); 112(7): 601-18.
- Mann S, Frasca K, Scherrer S, Henao-Martínez AF, Newman S, Ramanan P, et al. A Review of Leishmaniasis: Current Knowledge and Future Directions. Current Tropical Medicine Reports, (2021); 8(2):121-32.
- Omondi ZN, Arserim SK, Töz S, Özbel Y. Host–Parasite Interactions: Regulation of Leishmania Infection in Sand Fly. Acta Parasitologica, (2022); 67(2):606-18.
- Maksoud S, Ortega JT, Hidalgo M, Rangel HR. Leishmania donovani and HIV co-infection in vitro: Identification and characterization of main molecular players. Acta Tropica, (2022); 228:106248.
- Word Health Organization. Ending the neglect to attain the Sustainable Development Goals: a road map for neglected tropical diseases 2021-2030. Geneva: World Health Organization. Word Heal Organ, (2020);196.
- Scott P, Novais FO. Cutaneous leishmaniasis: immune responses in protection and pathogenesis. Nature Reviews Immunology, (2016); 16(9):581-92.
- Dorlo TPC, van Thiel PPAM, Huitema ADR, Keizer RJ, de Vries HJC, Beijnen JH, et al. Pharmacokinetics of Miltefosine in Old World Cutaneous Leishmaniasis Patients. Antimicrobial Agents and Chemotherapy, (2008); 52(8):2855-60.
- Monge-Maillo B, Lopez-Velez R. Miltefosine for Visceral and Cutaneous Leishmaniasis: Drug Characteristics and Evidence-Based Treatment Recommendations. Clinical Infectious Diseases, (2015); 60(9): 1398-404.
- Ajijur R, Salman A, Ahmad KMK. Combinatorial Design to Decipher Novel Lead Molecule against Mycobacterium tuberculosis. Biointerface Research in Applied Chemistry, (2021); 11(5):12993-3004.
- Khan MKA, Pokharkar NB, Al-Khodairy FM, Al-Marshad FM, Arif JM. Structural Perspective on Molecular Interaction of IgG and IgA with Spike and Envelope Proteins of SARS-CoV-2 and Its Implications to Non-Specific Immunity. Biointerface Research in Applied Chemistry, (2020); 11(3):10923-39.
- Khan MKA, Ahmad S, Rabbani G, Shahab U, Khan MS. Target‐based virtual screening, computational multiscoring docking and molecular dynamics simulation of small molecules as promising drug candidate affecting kinesin‐like protein KIFC1. Cell Biochemistry & Function, (2022); 40(5):451-472.
- Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics, (2011); 27(3):343-50.
- Alwabli AS. Lead Identification against 3C-like Protease of SARS-CoV-2 Via Target-based Virtual Screening and Molecular Dynamics Simulation. Journal of Young Pharmacists, (2022); 14(2):179-86.
- Subramanya HS, Bird LE, Brannigan JA, Wigley DB. Crystal structure of a DExx box DNA helicase. Nature (1996); 384(6607):379-83.
- Studer G, Rempfer C, Waterhouse AM, Gumienny R, Haas J, Schwede T. QMEANDisCo-distance constraints applied on model quality estimation. Elofsson A, editor. Bioinformatics, (2020); 36(6):1765-71.
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. Journal of Computational Chemistry, (1983); 4(2):187-217.
- Ahmad KMK, Salman A, Al-Khodairy Salman F, Al-Marshad Feras M, Alshahrani Abdulrahman M, Arif Jamal M. Computational Exploration of Dibenzo [a,l] Pyrene Interaction to DNA and its Bases: Possible Implications to Human Health. Biointerface Research in Applied Chemistry, (2020); 11(4):11272-83.
- Kiss R, Sandor M, Szalai FA. A public web service for drug discovery. Journal of Cheminformatics, (2012); 4(S1):P17.
- Shakil S. Molecular interaction of investigational ligands with human brain acetylcholinesterase. Journal of Cellular Biochemistry, (2019);120(7):11820-30.
- Ware JM, O’Connell EM, Brown T, Wetzler L, Talaat KR, Nutman TB, et al. Efficacy and Tolerability of Miltefosine in the Treatment of Cutaneous Leishmaniasis. Clinical Infectious Diseases, (2021);73(7):e2457-562.
- Castro M del M, Gomez MA, Kip AE, Cossio A, Ortiz E, Navas A, et al. Pharmacokinetics of Miltefosine in Children and Adults with Cutaneous Leishmaniasis. Antimicrobial Agents and Chemotherapy, (2017); 61(3): e02198-16.
- Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, et al. (2009). CHARMM: The biomolecular simulation program. Journal of Computational Chemistry, (2009); 30(10):1545-614.
- OLEG TROTT AJO, Schroer A. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. Journal of Computational Chemistry, (2010); 31(16):2967-70.
- Khan MKA, Akhtar S, Arif JM. Development of In Silico Protocols to Predict Structural Insights into the Metabolic Activation Pathways of Xenobiotics. Interdisciplinary Sciences: Computational Life Sciences, (2018;10(2):329-45.
- Khan MKA, Akhtar S, Arif JM. (2018). Structural Insight into the Mechanism of Dibenzo[a,l]pyrene and Benzo[a]pyrene-Mediated Cell Proliferation Using Molecular Docking Simulations. Interdisciplinary Sciences: Computational Life Sciences, (2018); 10(4):653-73.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, (2017); 7(1):42717.
- Attique S, Hassan M, Usman M, Atif R, Mahboob S, Al-Ghanim K, et al. A Molecular Docking Approach to Evaluate the Pharmacological Properties of Natural and Synthetic Treatment Candidates for Use against Hypertension. International Journal of Environmental Research and Public Health, (2019); 16(6):923.
- Egan WJ, Merz, KM, Baldwin JJ. Prediction of Drug Absorption Using Multivariate Statistics. Journal of Medicinal Chemistry, (2000), 43(21):3867-77.
- Egan WJ, Lauri G. Prediction of intestinal permeability. Advanced Drug Delivery Reviews (2002); 54(3):273-89.
- Baell JB, Holloway GA. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. Journal of Medicinal Chemistry,(2010); 53(7):2719-40.
- Brenk R, Schipani A, James D, Krasowski A, Gilbert IH, Frearson J, et al. Lessons Learnt from Assembling Screening Libraries for Drug Discovery for Neglected Diseases. ChemMedChem, (2008); 3(3):435-44.
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: Fast, flexible, and free. Journal of Computational Chemistry, (2005); 26(16):1701-18.
- Vanommeslaeghe K, MacKerell AD. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. Journal of Chemical Information and Modeling, (2012); 52(12):3144-54.
- Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. Journal of Computational Chemistry, (2010); 31(4):671-90.
- Petersen HG. Accuracy and efficiency of the particle mesh Ewald method. Journal of Chemical Physics, (1995); 103(9):3668-79.
- Stenberg S, Stenqvist B. An Exact Ewald Summation Method in Theory and Practice. Journal of Physical Chemistry,(2020); 124(19):3943-6.
- Fischer NM, van Maaren PJ, Ditz JC, Yildirim A, van der Spoel D. Properties of Organic Liquids when Simulated with Long-Range Lennard-Jones Interactions. Journal of Chemical Theory and Computation, (2015); 11(7):2938-44.
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography, (1993); 26(2):283-91.
- Şahin S, Dege N. A newly synthesized small molecule: the evaluation against Alzheimer’s Disease by in silico drug design and computational structure analysis methods. Journal of Molecular Structure, (2021); 1236(1):130337.
- Ghose AK, Viswanadhan VN, Wendoloski JJ. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. Journal of Combinatorial Chemistry, (1999); 1(1):55-68.
- Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. Journal of Medicinal Chemistry, (2002); 45(12):2615-23.
- Muegge I, Heald SL, Brittelli D. Simple Selection Criteria for Drug-like Chemical Matter. Journal of Medicinal Chemistry,(2001); 44(12):1841-6.
- Martin YC. A Bioavailability Score. ournal of Medicinal Chemistry, (2005); 48(9):3164-70.
- Kausar MA, Shahid S, Anwar S, Kuddus M, Khan MKA, Alotaibi AD, et al. Identifying Natural Therapeutics against Diabetes via Inhibition of Dipeptidyl Peptidase 4: Molecular Docking and MD Simulation Study. Indian Journal of Pharmaceutical Education and Research, (2022); 56(1s):s21-31.
- Kausar MA, Shahid S, Anwar S, Kuddus M, Khan MKA, Khalifa AM, et al. Identifying the alpha-glucosidase inhibitory potential of dietary phytochemicals against diabetes mellitus type 2 via molecular interactions and dynamics simulation. Cellular and Molecular Biology, (2022); 67(5):16-26.
- Gupta R, Brosh RMJ. Helicases as Prospective Targets for Anti-Cancer Therapy. Anti-Cancer Agents in Medicinal Chemistry. (2008); 8 (4): 390-401.
- Singleton MR, Dillingham MS, Gaudier M, Kowalczykowski SC, Wigley DB. Crystal Structure of RecBCD Enzyme Reveals a Machine for Processing DNA Breaks. Nature (2004); 432 (7014): 187-193.
- Itsathitphaisarn O, Wing RA, Eliason WK, Wang J, Steitz TA. The Hexameric Helicase DnaB Adopts a Nonplanar Conformation during Translocation. Cell 2012, 151 (2): 267-277.
- Imanishi H, Sasaki YF, Matsumoto K, Watanabe M, Ohta T, Shirasu Y, Tutikawa K. Suppression of 6-TG-resistant mutations in V79 cells and recessive spot formations in mice by vanillin. Mutation Research, (1990); 243(2):151-8.
- Das S, Roy P, Mondal S, Bera T, Mukherjee A. One pot synthesis of gold nanoparticles and application in chemotherapy of wild and resistant type visceral leishmaniasis. Colloids Surf B Biointerfaces (2013); l 1:107:27-34.
- Singh PP, Chakraborty P. Malaria: autophagy as a potential therapeutic target. Journal of Pharmacy and Pharmacology, (2016); 4:298-306.
This work is licensed under a Creative Commons Attribution-Non Commercial 4.0 International License. To read the copy of this license please visit: https://creativecommons.org/licenses/by-nc/4.0